
Four Novel Disease-Causing Variants in the NOTCH3 Gene in Russian Patients with CADASIL

 , ,
, ,
Abstract
:1. Background
2. Materials and Methods
2.1. Patients
2.2. MRI Scan
2.3. Molecular Genetic Methods
2.4. Patients
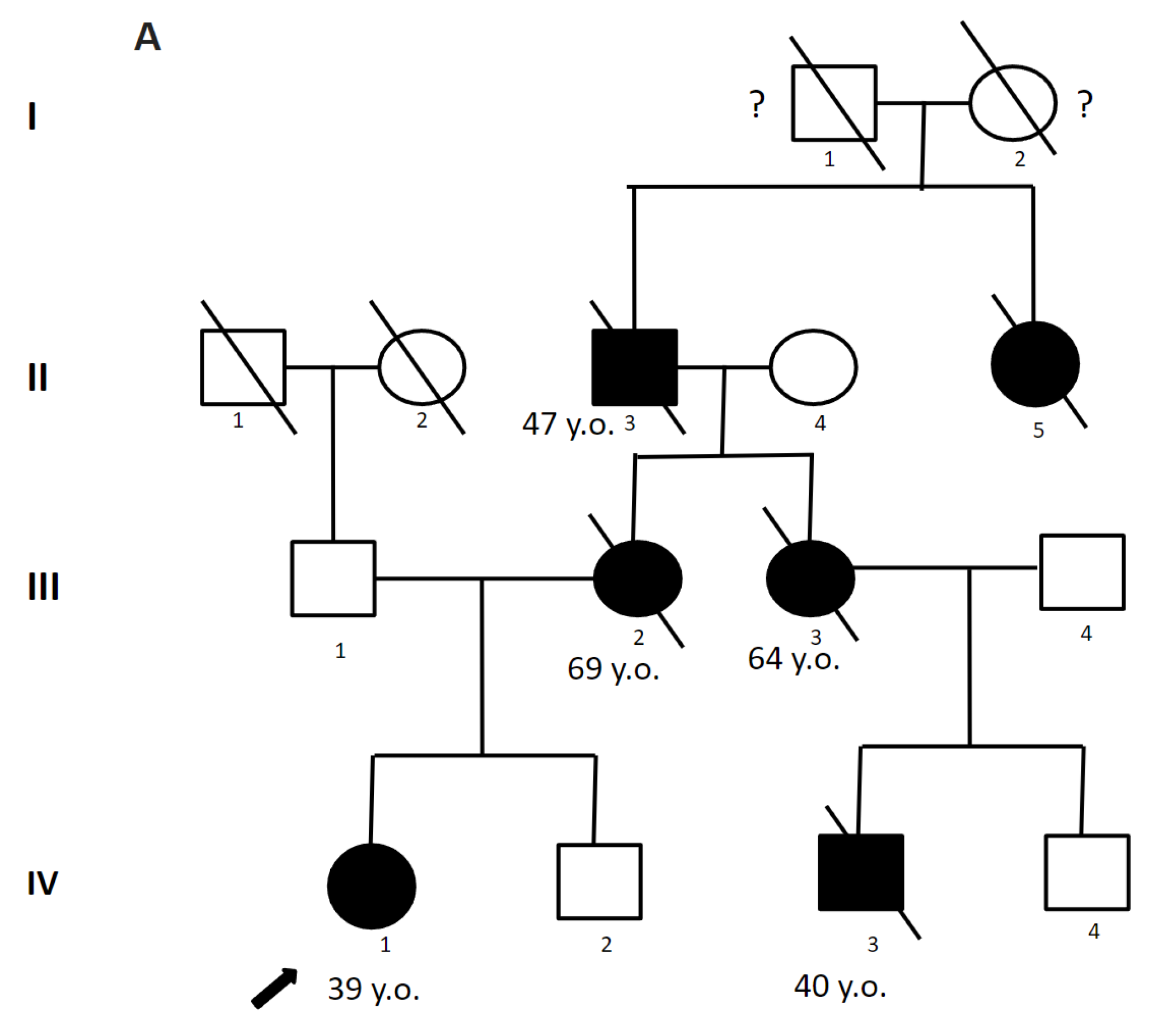
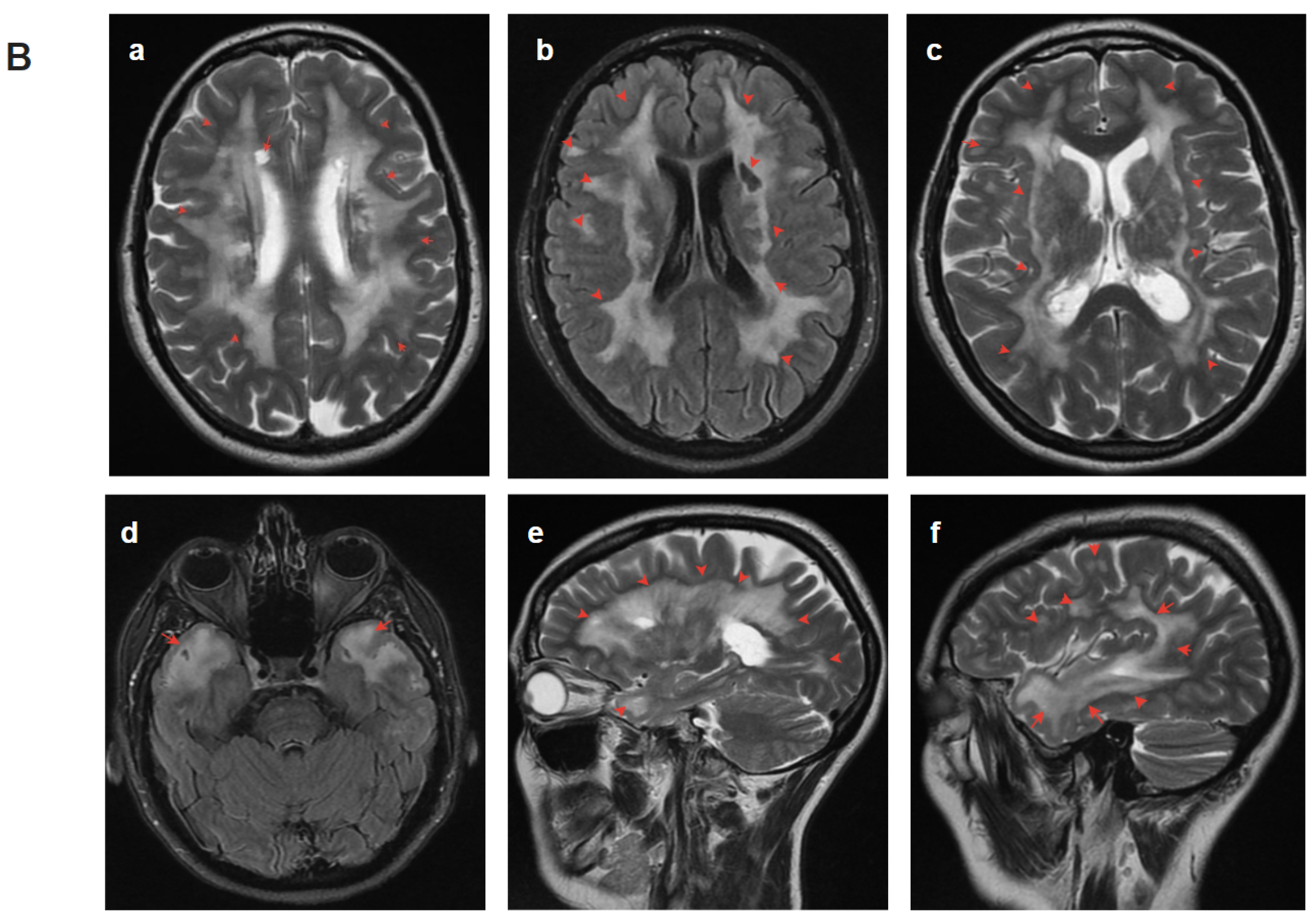
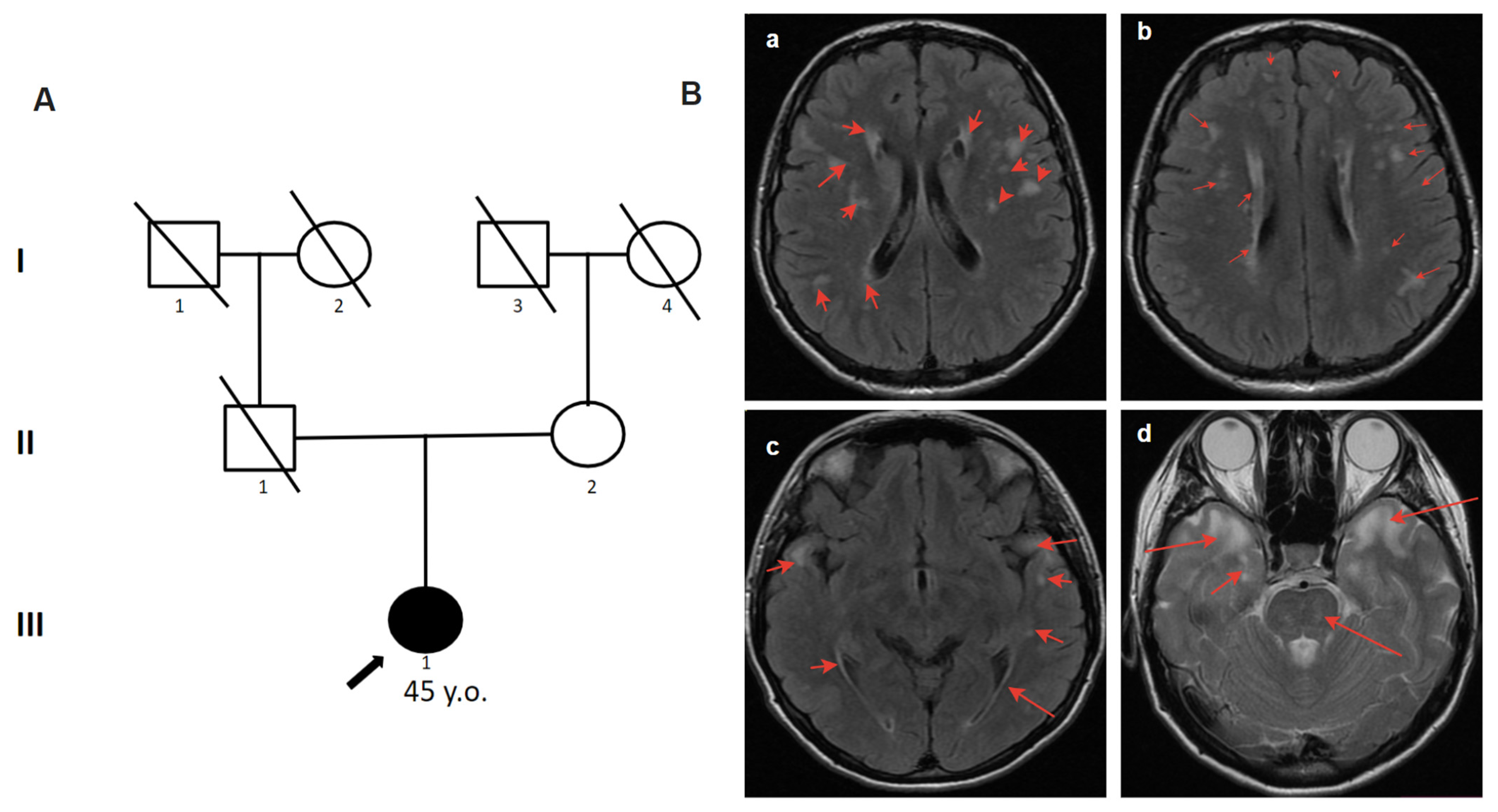
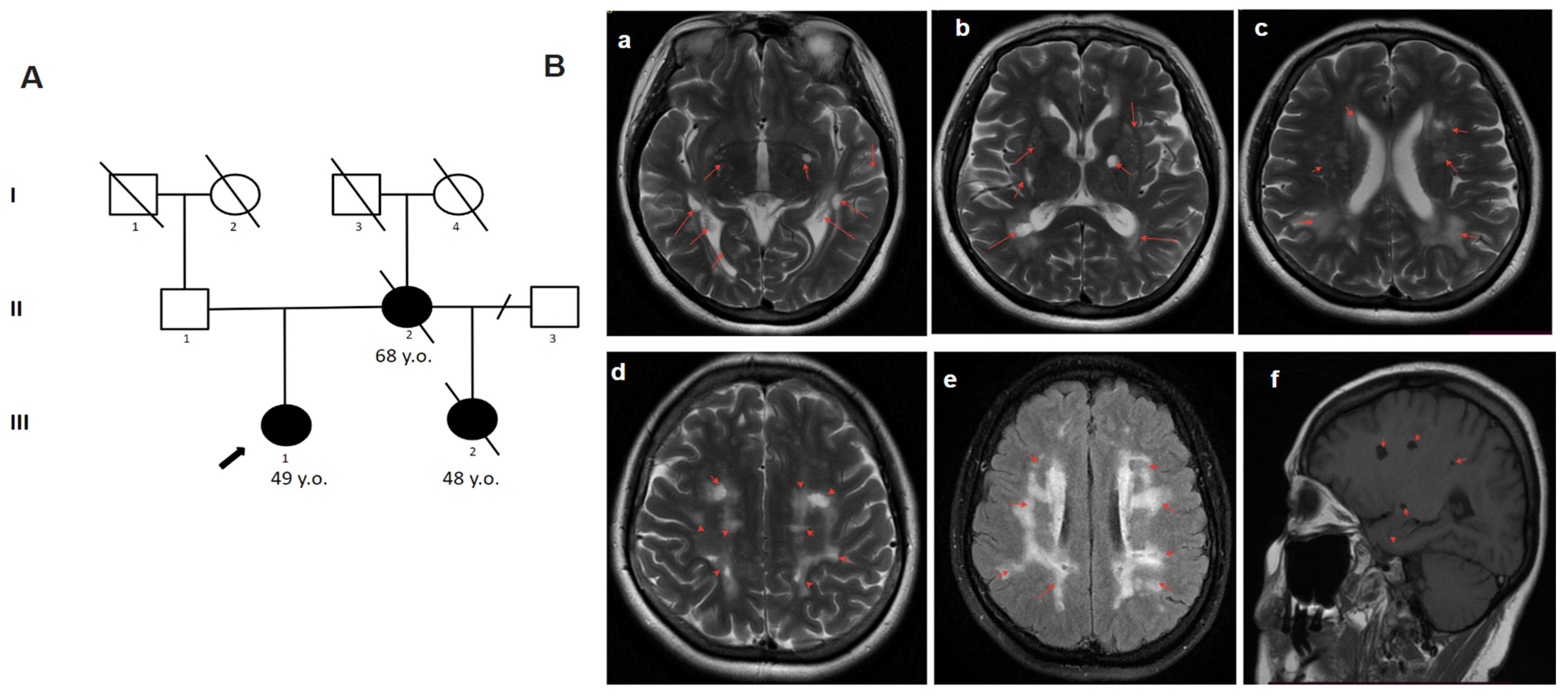
2.4.1. Patient 1
2.4.2. Patient 2
2.4.3. Patient 3
2.4.4. Patient 4.1
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CADASIL | cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| WMH | white matter hyperintensities |
| TIA | transient ischemic attack |
| CVA | cerebrovascular accident |
| MRI | magnetic resonance imaging |
| T1WI | T1-weighted images |
| T2WI | T2-weighted images |
| FLAIR | fluid-attenuated inversion recovery |
References
- André, C. CADASIL: Pathogenesis, clinical and radiological findings and treatment. Arq. Neuro-Psiquiatr. 2010, 68, 287–299. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2012, 493, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Danchenko, I.Y.; Kulesh, A.A.; Drobakha, V.E.; Kanivets, I.V.; Akimova, I.A.; Monak, A.A. CADASIL syndrome: Differential diagnosis with multiple sclerosis. Zhurnal Nevrologii i Psikhiatrii imeni SS Korsakova 2019, 119, 128–136. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.-G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Rutten, J.W.; Van Eijsden, B.J.; Duering, M.; Jouvent, E.; Opherk, C.; Pantoni, L.; Federico, A.; Dichgans, M.; Markus, H.S.; Chabriat, H.; et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet. Med. 2018, 21, 676–682. [Google Scholar] [CrossRef]
- Schoemaker, D.; Quiroz, Y.T.; Torrico-Teave, H.; Arboleda-Velasquez, J.F. Clinical and research applications of magnetic resonance imaging in the study of CADASIL. Neurosci. Lett. 2019, 698, 173–179. [Google Scholar] [CrossRef]
- Dichgans, M.; Mayer, M.; Uttner, I.; Brüning, R.; Müller-Höcker, J.; Rungger, G.; Ebke, M.; Klockgether, T.; Gasser, T. The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann. Neurol. 1998, 44, 731–739. [Google Scholar] [CrossRef]
- Opherk, C.; Peters, N.; Herzog, J.; Luedtke, R.; Dichgans, M. Long-term prognosis and causes of death in CADASIL: A retrospective study in 411 patients. Brain 2004, 127, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- Liem, M.K.; Oberstein, S.A.L.; van der Grond, J.; Ferrari, M.D.; Haan, J. CADASIL and migraine: A narrative review. Cephalalgia 2010, 30, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M. Cadasil. In Handbook of Clinical Neurology; Elsevier: Ann Arbor, MI, USA, 2018; Volume 148, pp. 733–743. [Google Scholar] [CrossRef]
- Chabriat, H.; Vahedi, K.; Bousser, M.; Iba-Zizen, M.; Joutel, A.; Nibbio, A.; Nagy, T.; Lasserve, E.T.; Krebs, M.; Julien, J.; et al. Clinical spectrum of CADASIL: A study of 7 families. Lancet 1995, 346, 934–939. [Google Scholar] [CrossRef]
- O’Sullivan, M.; Jarosz, J.; Martin, R.; Deasy, N.; Powell, J.; Markus, H. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 2001, 56, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Genetic Cause of Cerebral Small Vessel Disease. J. Clin. Neurol. 2010, 6, 1–9. [Google Scholar] [CrossRef]
- Boom, R.v.D.; Oberstein, S.A.J.L.; Ferrari, M.D.; Haan, J.; van Buchem, M.A. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: MR Imaging Findings at Different Ages—3rd–6th Decades1. Radiology 2003, 229, 683–690. [Google Scholar] [CrossRef]
- Oberstein, S.A.J.L.; Boom, R.v.D.; Middelkoop, H.A.M.; Ferrari, M.D.; Knaap, Y.M.; van Houwelingen, H.C.; Breuning, M.H.; van Buchem, M.A.; Haan, J. Incipient CADASIL. Arch. Neurol. 2003, 60, 707–712. [Google Scholar] [CrossRef]
- Golomb, M.R.; Sokol, D.K.; Walsh, L.E.; Christensen, C.K.; Garg, B.P. Recurrent hemiplegia, normal MRI, and NOTCH3 mutation in a 14-year-old: Is this early CADASIL? Neurology 2004, 62, 2331–2332. [Google Scholar] [CrossRef]
- Samões, R.; Alves, J.E.; Taipa, R.; Silva, J.; Pires, M.M.; Pereira-Monteiro, J.M. CADASIL: MRI may be normal in the fourth decade of life—A case report. Cephalalgia 2016, 36, 1082–1085. [Google Scholar] [CrossRef]
- Zhu, S.; Nahas, S.J. CADASIL: Imaging Characteristics and Clinical Correlation. Curr. Pain Headache Rep. 2016, 20, 57. [Google Scholar] [CrossRef]
- Duchesnay, E.; Selem, F.H.; De Guio, F.; Dubois, M.; Mangin, J.-F.; Duering, M.; Ropele, S.; Schmidt, R.; Dichgans, M.; Chabriat, H.; et al. Different Types of White Matter Hyperintensities in CADASIL. Front. Neurol. 2018, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, G.E. Cadasil. In Encyclopedia of the Neurological Sciences; Academic Press: Cambridge, MA, USA, 2017; pp. 495–496. [Google Scholar] [CrossRef]
- Tikka, S.; Mykkänen, K.; Ruchoux, M.-M.; Bergholm, R.; Junna, M.; Pöyhönen, M.; Yki-Järvinen, H.; Joutel, A.; Viitanen, M.; Baumann, M.; et al. Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 2009, 132, 933–939. [Google Scholar] [CrossRef]
- Morroni, M.; Marzioni, D.; Ragno, M.; Di Bella, P.; Cartechini, E.; Pianese, L.; Lorenzi, T.; Castellucci, M.; Scarpelli, M. Role of Electron Microscopy in the Diagnosis of Cadasil Syndrome: A Study of 32 Patients. PLoS ONE 2013, 8, e65482. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Nakajima, M.; Misumi, Y.; Nakahara, K.; Shinriki, S.; Tasaki, M.; Matsui, H.; Ueda, M. Detection of Vascular NOTCH3 Deposits in Unfixed Frozen Skin Biopsy Sample in CADASIL. Front. Neurol. 2022, 13, 881528. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Family Number | Gender | Nucleotide Variants | Amino Acid Change | Exon/Intron Number | EGFr Domains | AGMG Classification | Evidence of Pathogenicity | Program |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | f. | c.208G>T | p.Gly70Cys | 1 exon | 1 | Likely pathogenic | PM2, PP3, PM1, PP4 | MutPred, DEOGEN2, M-CAP, Mutation assessor, MVP, FATHMM, FATHMM-MKL, PROVEAN |
| 2 | 2 | f. | c.341-1G>C | - | 3 intron | - | Pathogenic | PM2, PVS1, PP3, PP4 | SpliceAI, SPiP, MMSplice |
| 3 | 3 | f. | c.1136G>A | p.Cys379Tyr | 7 exon | 9 | Likely pathogenic | PM2, PM5, PP3, PM1, PP4 | SIFT, SIFT4G, Polyphen2_HDIV, Polyphen2_HVAR, FATHMM, PROVEAN, DEOGEN2 |
| 4.1 | 4 | m. | c.1547G>A | p.Cys516Tyr | 10 exon | 13 | Pathogenic | PM1, PM5, PP5, PM2, PP3 | MutPred, DEOGEN2, M-CAP, Mutation assessor, MVP, FATHMM, FATHMM-MKL, PROVEAN, EIGEN, EIGEN PC, LIST-S2, FATHMM-XF, PrimateAI, SIFT, SIFT4G |
| 4.2 | 4 | m. | c.1547G>A | p.Cys516Tyr | 10 exon | 13 | Pathogenic | PM2, PP3, PP1, PP4, PM5, PM1 | MutPred, DEOGEN2, M-CAP, Mutation assessor, MVP, FATHMM, FATHMM-MKL, PROVEAN, EIGEN, EIGEN PC, LIST-S2, FATHMM-XF, PrimateAI, SIFT, SIFT4G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bostanova, F.; Tsygankova, P.; Nagornov, I.; Dadali, E.; Bessonova, L.; Kulesh, A.; Drobakha, V.; Danchenko, I.; Kanivets, I.; Zakharova, E. Four Novel Disease-Causing Variants in the NOTCH3 Gene in Russian Patients with CADASIL. Genes 2023, 14, 1715. https://doi.org/10.3390/genes14091715
Bostanova F, Tsygankova P, Nagornov I, Dadali E, Bessonova L, Kulesh A, Drobakha V, Danchenko I, Kanivets I, Zakharova E. Four Novel Disease-Causing Variants in the NOTCH3 Gene in Russian Patients with CADASIL. Genes. 2023; 14(9):1715. https://doi.org/10.3390/genes14091715
Chicago/Turabian StyleBostanova, Fatima, Polina Tsygankova, Ilya Nagornov, Elena Dadali, Lyudmila Bessonova, Aleksey Kulesh, Viktor Drobakha, Irina Danchenko, Ilya Kanivets, and Ekaterina Zakharova. 2023. "Four Novel Disease-Causing Variants in the NOTCH3 Gene in Russian Patients with CADASIL" Genes 14, no. 9: 1715. https://doi.org/10.3390/genes14091715
APA StyleBostanova, F., Tsygankova, P., Nagornov, I., Dadali, E., Bessonova, L., Kulesh, A., Drobakha, V., Danchenko, I., Kanivets, I., & Zakharova, E. (2023). Four Novel Disease-Causing Variants in the NOTCH3 Gene in Russian Patients with CADASIL. Genes, 14(9), 1715. https://doi.org/10.3390/genes14091715