
Proteomics as a Metrological Tool to Evaluate Genome Annotation Accuracy Following De Novo Genome Assembly: A Case Study Using the Atlantic Bottlenose Dolphin (Tursiops truncatus)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Source and Preparation
2.2. Mass Spectrometry
2.3. Protein Search Parameters
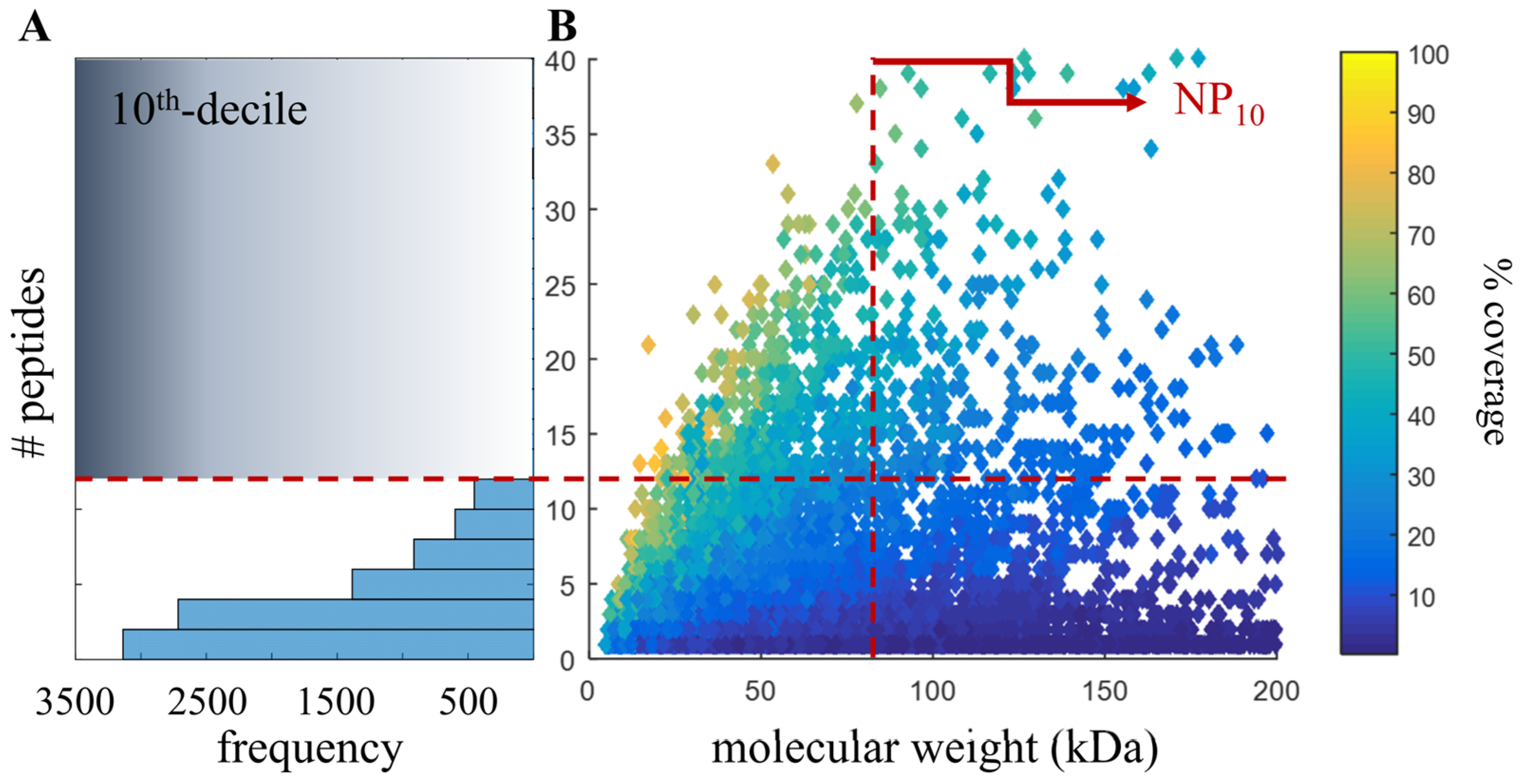
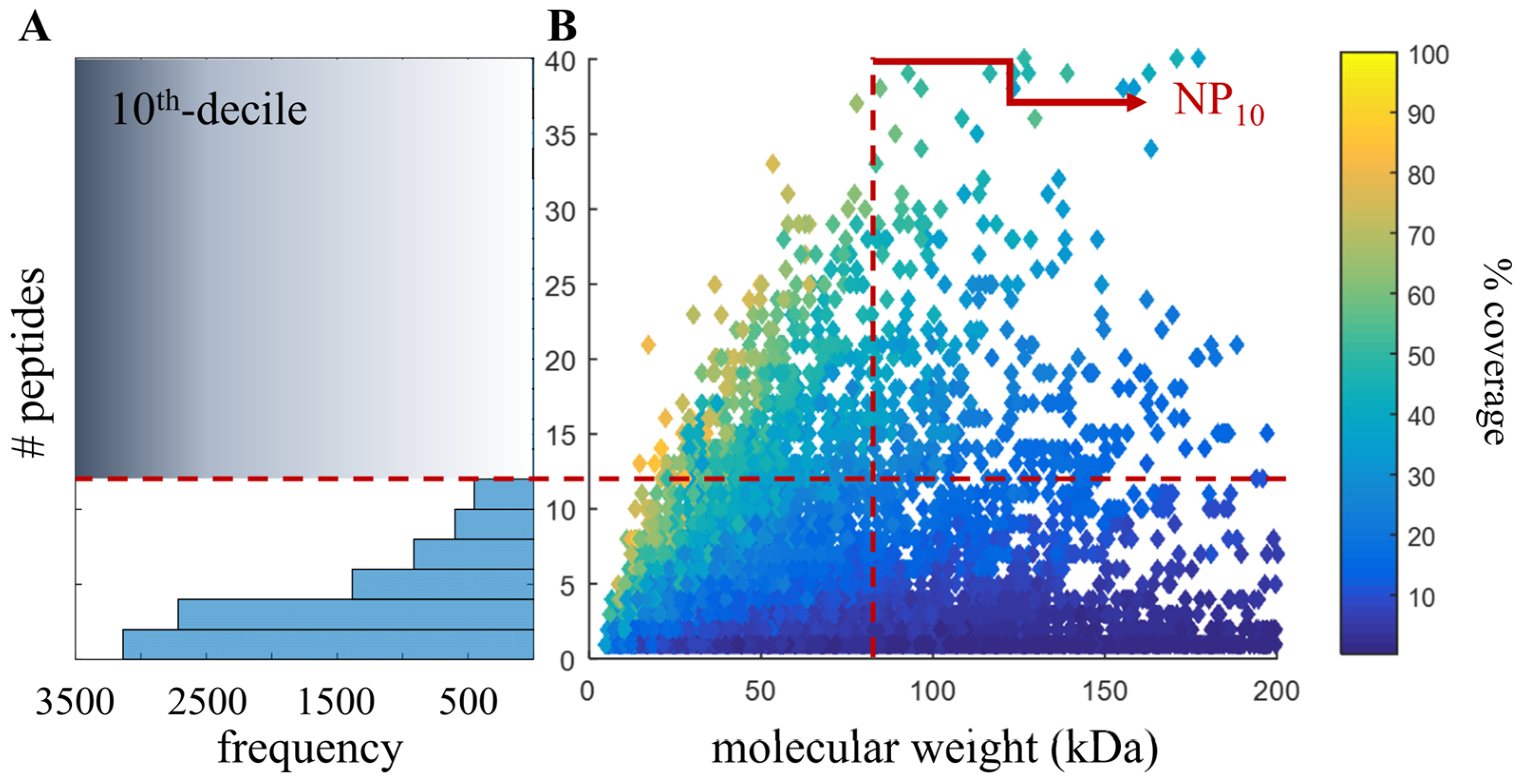
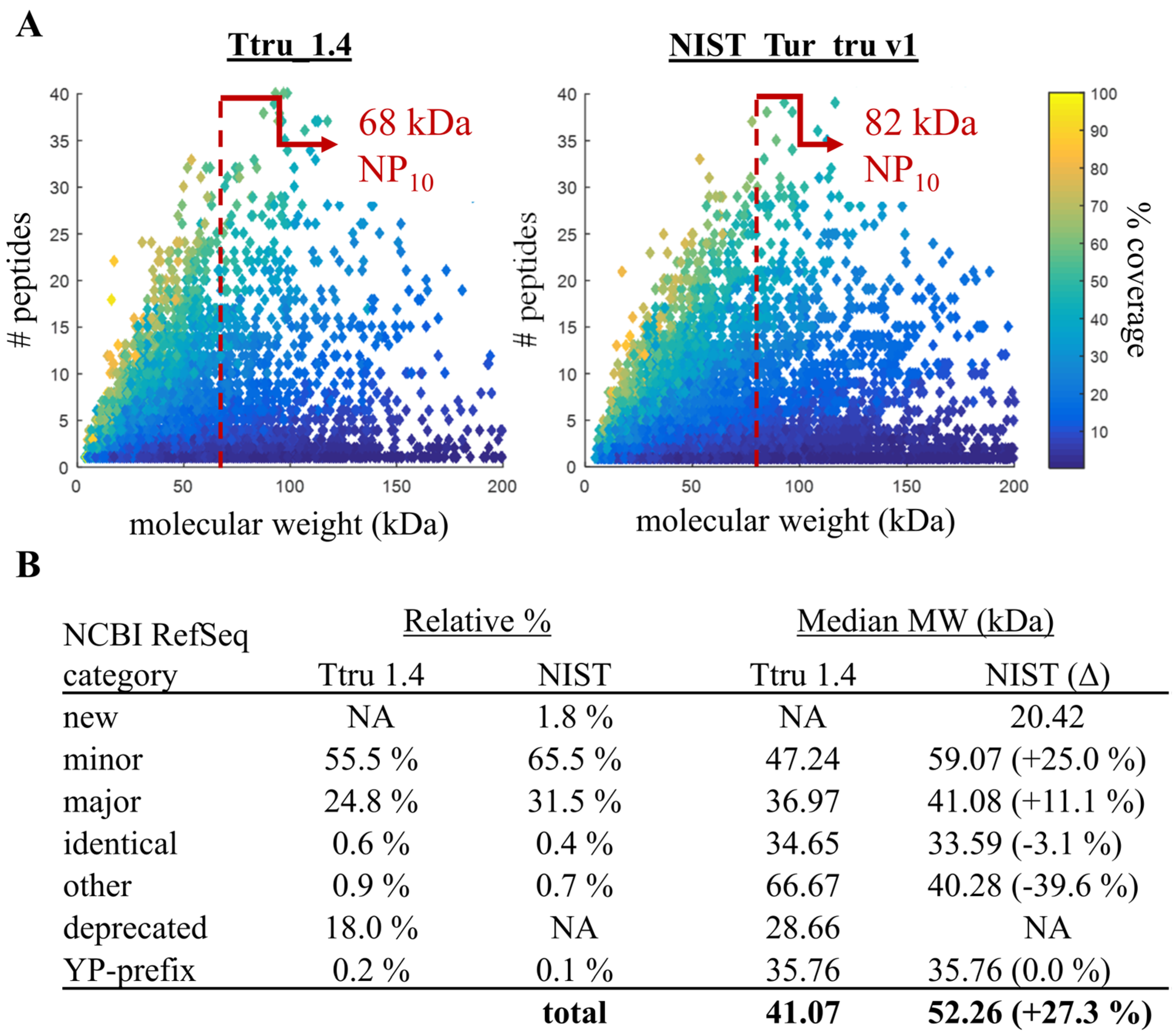
2.4. Proteomic-Based Quality Metric for Annotation Quality
2.5. Details of Online Resources from NCBI
3. Results
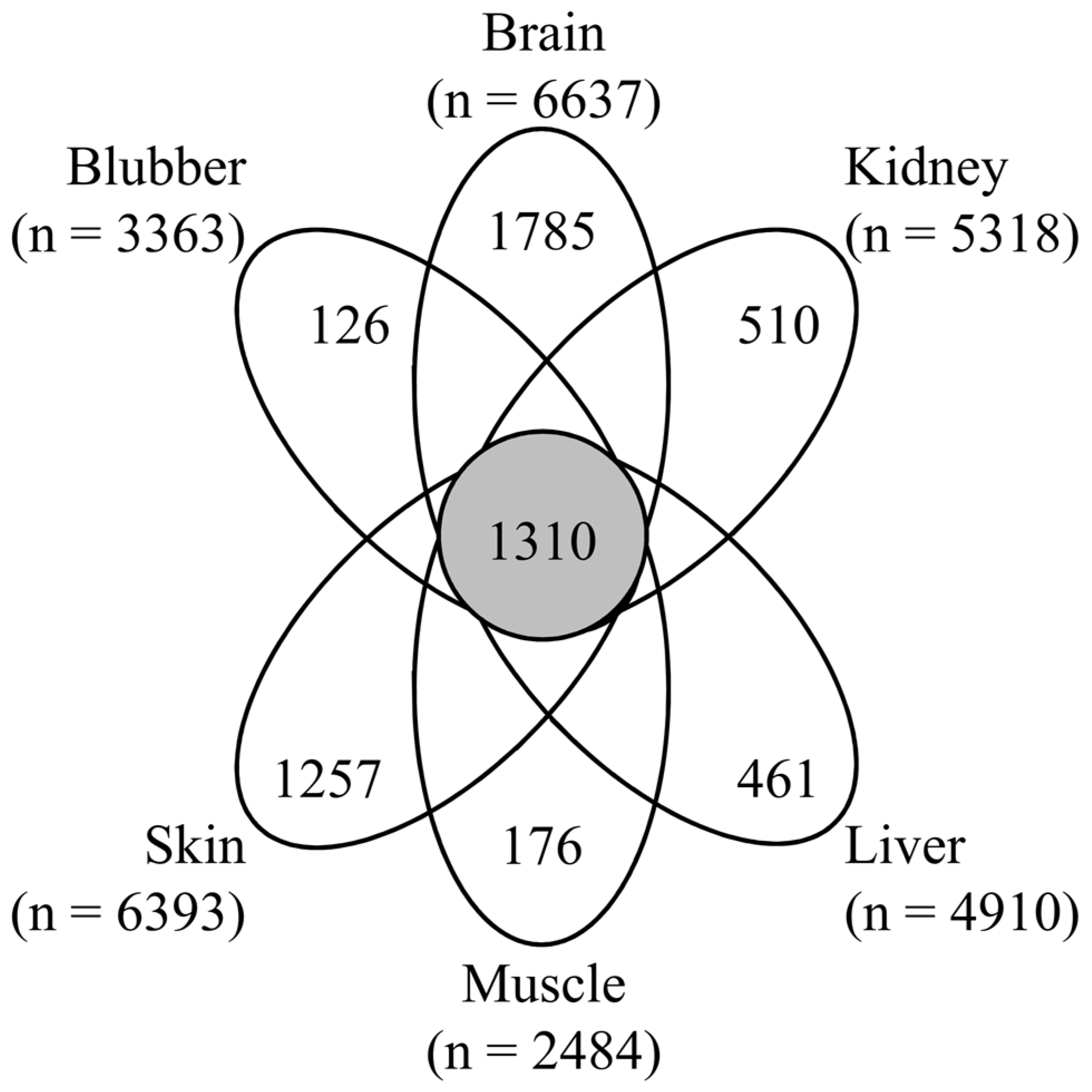
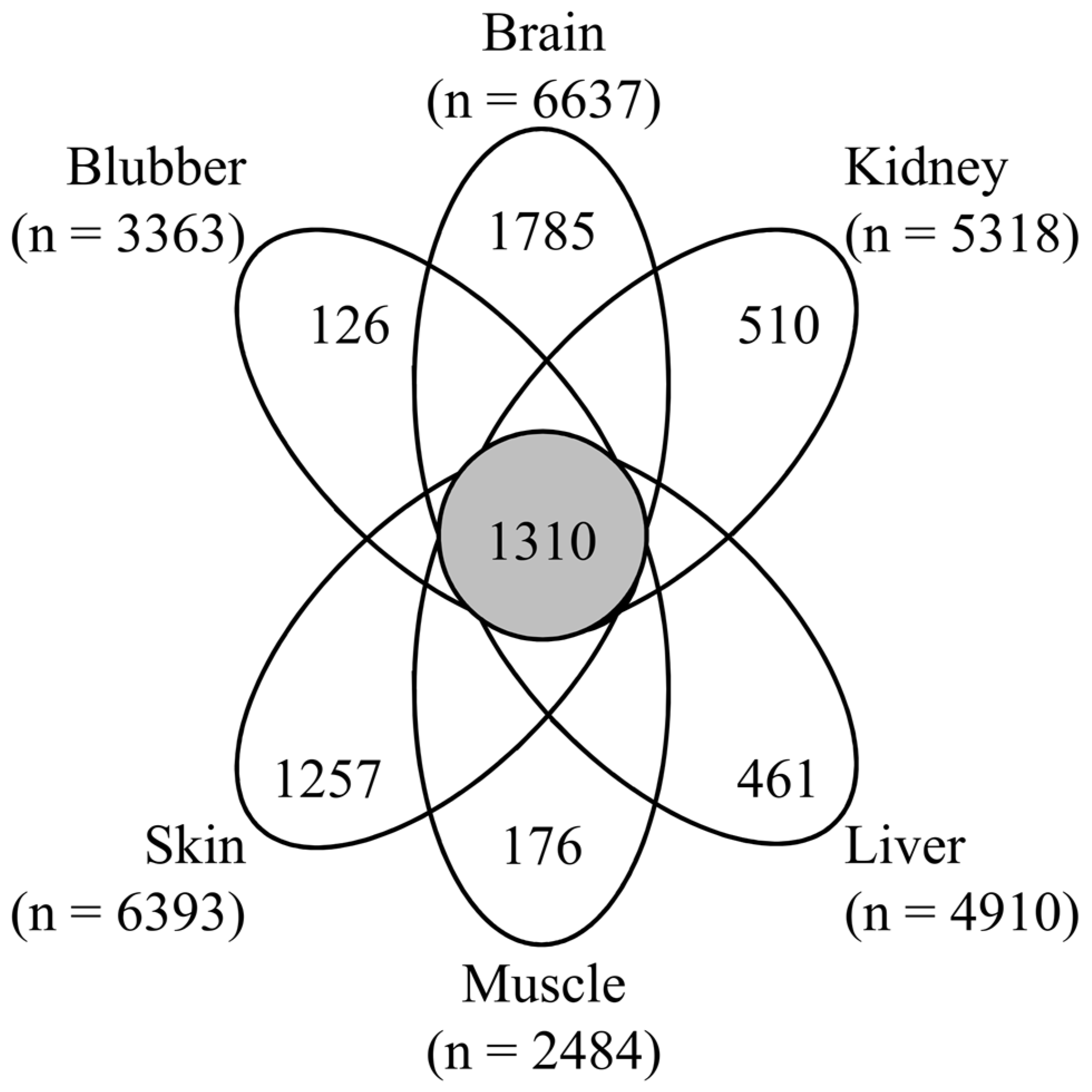
3.1. Proteomic Analysis of Six Tissues Using NIST_Tur_tru v1
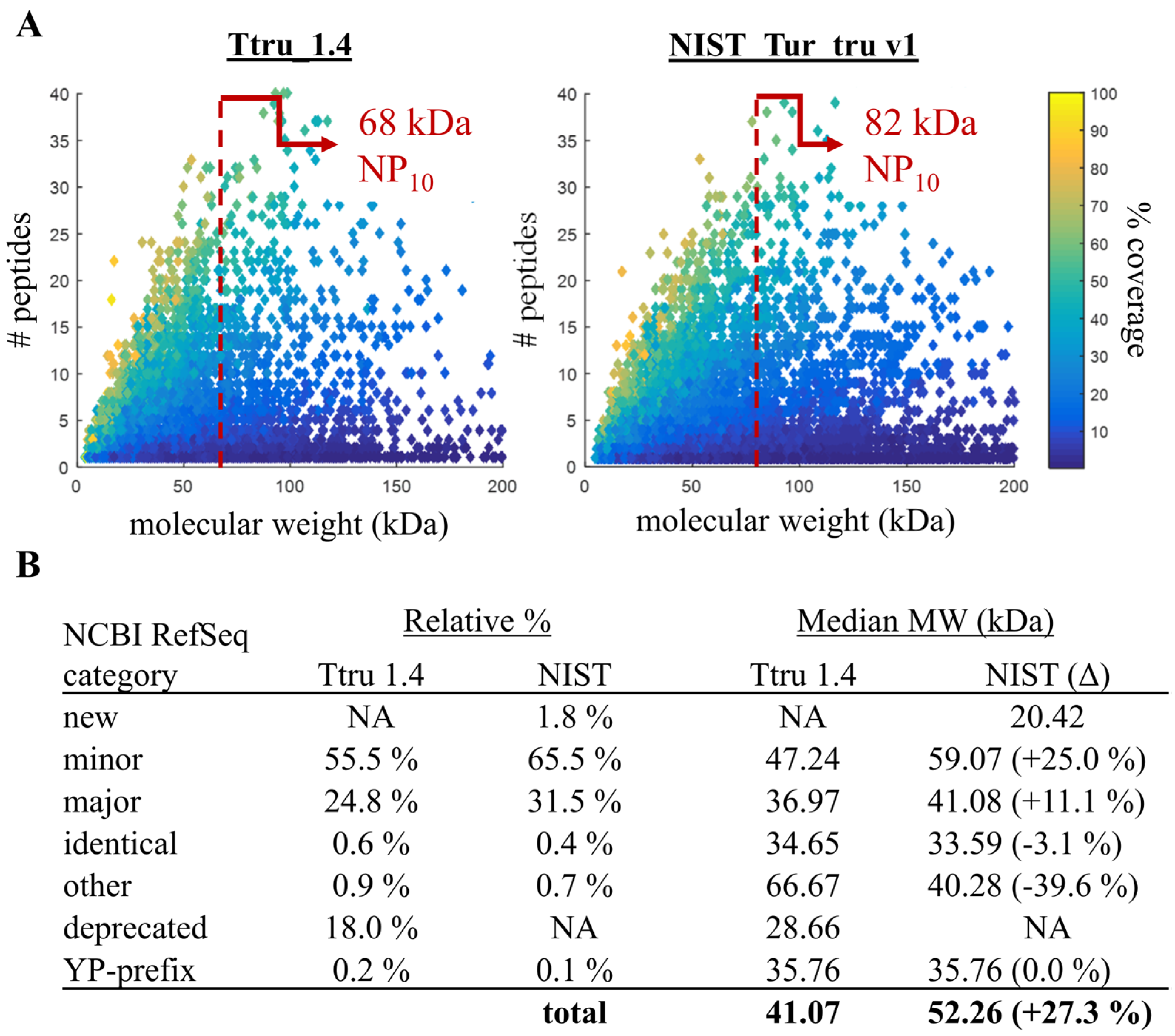
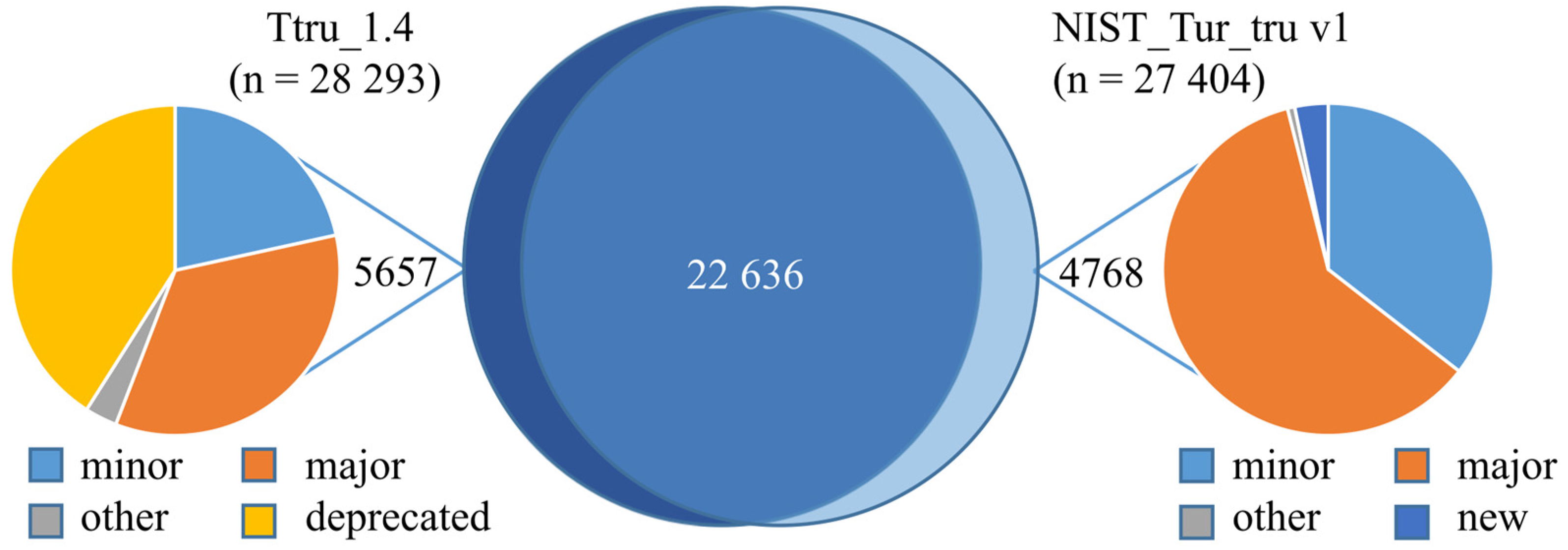
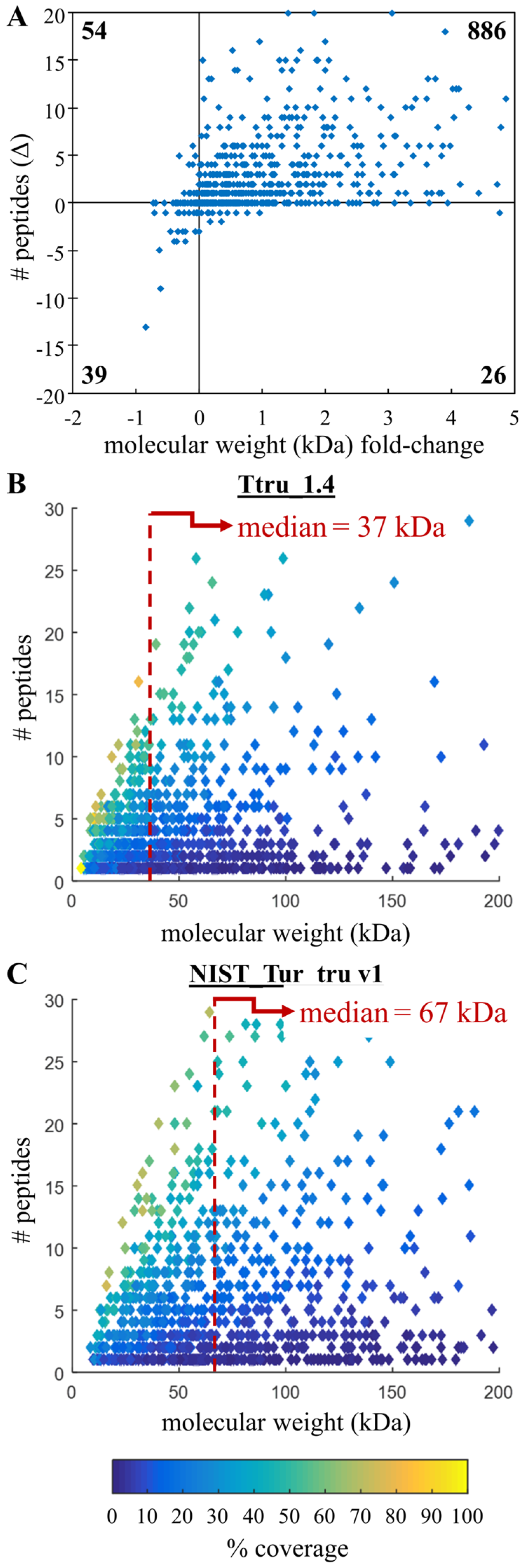
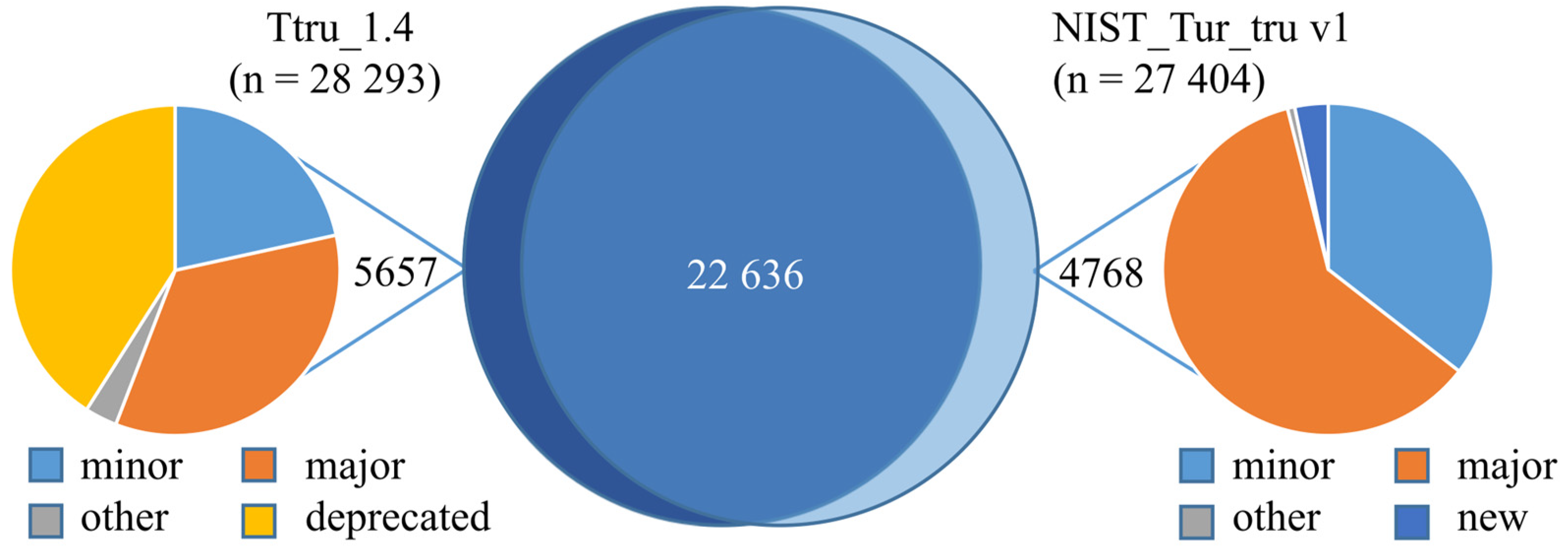
3.2. Comparison of Ttru_1.4 and NIST_Tur_tru v1
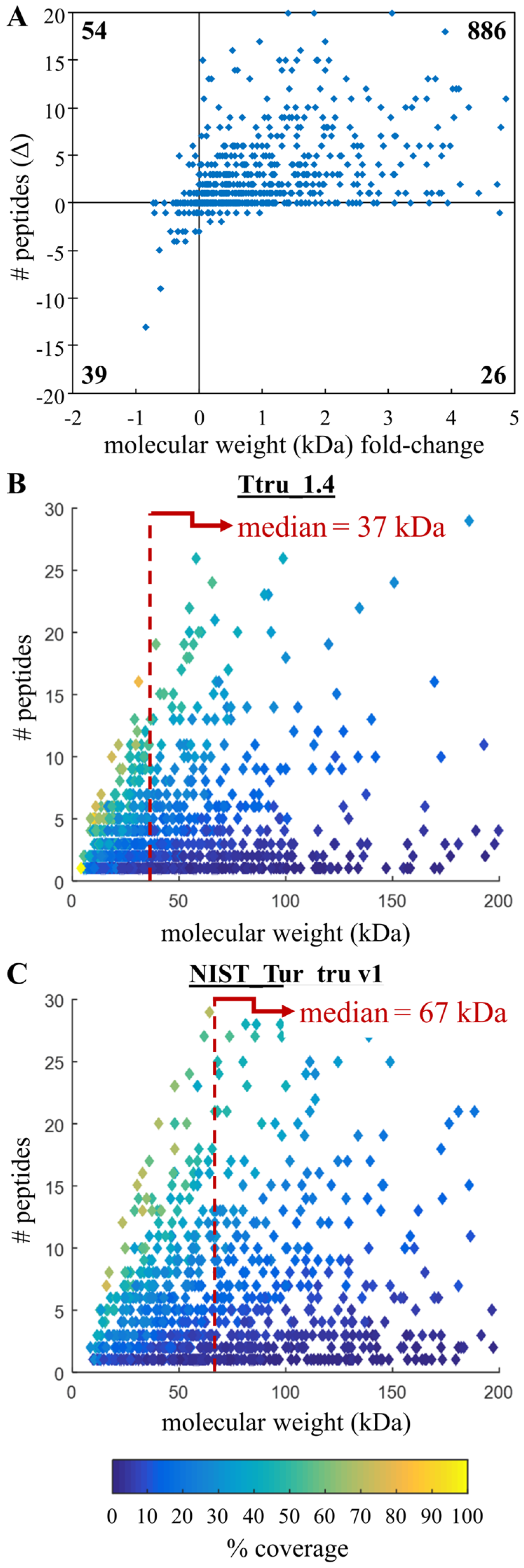
3.3. Confirming Improvements in Gene Annotation
3.4. Comparing Peptide Identifications
3.5. Specific Examples of Annotation Improvements
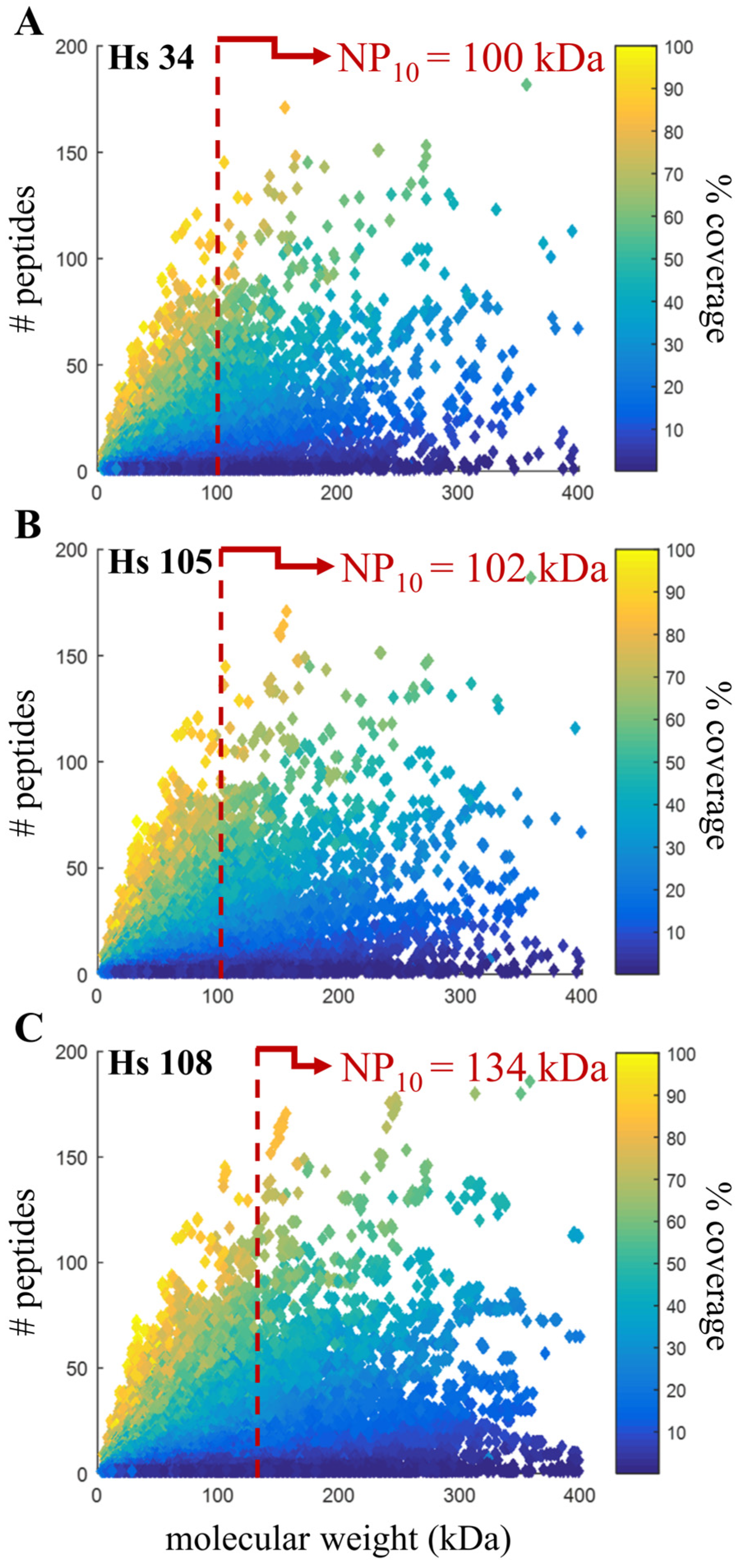
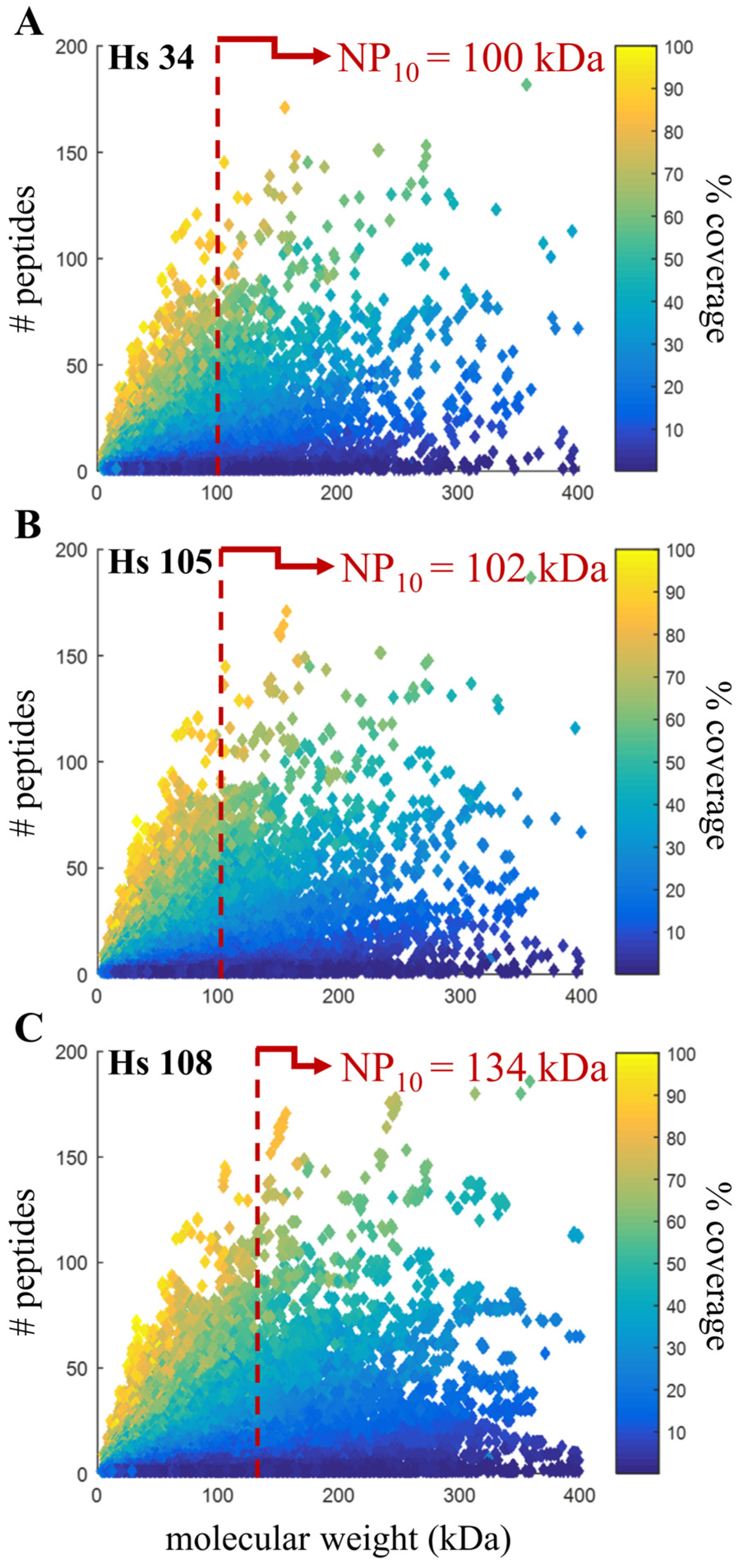
3.6. Confirming Quality Metric in Human Annotations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: https://www.genome.gov/sequencingcostsdata (accessed on 18 August 2023).
- Heck, M.; Neely, B.A. Proteomics in Non-model Organisms: A New Analytical Frontier. J. Proteome Res. 2020, 19, 3595–3606. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef]
- Worley, K.C. A golden goat genome. Nat. Genet. 2017, 49, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.W.; Naguib, A.; Weisenfeld, N.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.; Scott, A.F. Improved de novo Genome Assembly: Linked-Read Sequencing Combined with Optical Mapping Produce a High Quality Mammalian Genome at Relatively Low Cost. bioRxiv 2017, 128348. [Google Scholar] [CrossRef]
- Dudchenko, O.; Shamim, M.S.; Batra, S.S.; Durand, N.C.; Musial, N.T.; Mostofa, R.; Pham, M.; Glenn St Hilaire, B.; Yao, W.; Stamenova, E.; et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv 2018, 254797. [Google Scholar] [CrossRef]
- Richards, S. It’s More Than Stamp Collecting: How Genome Sequencing Can Unify Biological Research. Trends Genet. TIG 2015, 31, 411–421. [Google Scholar] [CrossRef]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Ning, Z.; Keane, T.; Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009, 37, 289–297. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Berkeley, M.; Kriventseva, E.V. OrthoDB in 2020: Evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2021, 49, D389–D393. [Google Scholar] [CrossRef]
- Lam, H.; Deutsch, E.W.; Eddes, J.S.; Eng, J.K.; Stein, S.E.; Aebersold, R. Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods 2008, 5, 873–875. [Google Scholar] [CrossRef]
- Burke, M.C.; Mirokhin, Y.A.; Tchekhovskoi, D.V.; Markey, S.P.; Heidbrink Thompson, J.; Larkin, C.; Stein, S.E. The Hybrid Search: A Mass Spectral Library Search Method for Discovery of Modifications in Proteomics. J. Proteome Res. 2017, 16, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Burke, M.; Mirokhin, Y.A.; Tchekhovskoi, D.V.; Markey, S.P.; Yu, W.; Chaerkady, R.; Hess, S.; Stein, S.E. Reverse and Random Decoy Methods for False Discovery Rate Estimation in High Mass Accuracy Peptide Spectral Library Searches. J. Proteome Res. 2018, 12, 846–857. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e584. [Google Scholar] [CrossRef]
- Sinitcyn, P.; Richards, A.L.; Weatheritt, R.J.; Brademan, D.R.; Marx, H.; Shishkova, E.; Meyer, J.G.; Hebert, A.S.; Westphall, M.S.; Blencowe, B.J.; et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol. 2023, 1–11. [Google Scholar] [CrossRef]
- Johnson, R.S.; Searle, B.C.; Nunn, B.L.; Gilmore, J.M.; Phillips, M.; Amemiya, C.T.; Heck, M.; MacCoss, M.J. Assessing Protein Sequence Database Suitability Using De Novo Sequencing. Mol. Cell. Proteom. 2020, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Liu, Y.; Thomas, G.W.; Vinar, T.; Alfoldi, J.; Deng, J.; Dugan, S.; van Elk, C.E.; Hunter, M.E.; Joshi, V.; et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef]
- Sobolesky, P.; Parry, C.; Boxall, B.; Wells, R.; Venn-Watson, S.; Janech, M.G. Proteomic Analysis of Non-depleted Serum Proteins from Bottlenose Dolphins Uncovers a High Vanin-1 Phenotype. Sci. Rep. 2016, 6, 33879. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.S.; Becker, P.R.; Porter, B.J.; Ellisor, M.B.; Moors, A.J.; Wise, S.A. Design and Applications of the National Institute of Standards and Technology’s (NIST’s) Environmental Specimen Banking Programs. Cell Preserv. Technol. 2008, 6, 59–72. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Brown, G.R.; Hiatt, S.M.; Thibaud-Nissen, F.; Astashyn, A.; Ermolaeva, O.; Farrell, C.M.; Hart, J.; Landrum, M.J.; McGarvey, K.M.; et al. RefSeq: An update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–D763. [Google Scholar] [CrossRef] [PubMed]
- NCBI RefSeq. Available online: Ftp://ftp.ncbi.nih.gov/genomes/refseq/ (accessed on 18 August 2023).
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef]
- Jones, S.J.; Taylor, G.A.; Chan, S.; Warren, R.L.; Hammond, S.A.; Bilobram, S.; Mordecai, G.; Suttle, C.A.; Miller, K.M.; Schulze, A. The Genome of the Beluga Whale (Delphinapterus leucas). Genes 2017, 8, 378. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef]
- NCBI. Tursiops Truncatus Annotation Release 101 Annotation Report. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Tursiops_truncatus/101/ (accessed on 18 August 2023).
- Shamsi, A.; Bano, B. Journey of cystatins from being mere thiol protease inhibitors to at heart of many pathological conditions. Int. J. Biol. Macromol. 2017, 102, 674–693. [Google Scholar] [CrossRef]
- Neely, B.A.; Carlin, K.P.; Arthur, J.M.; McFee, W.E.; Janech, M.G. Ratiometric Measurements of Adiponectin by Mass Spectrometry in Bottlenose Dolphins (Tursiops truncatus) with Iron Overload Reveal an Association with Insulin Resistance and Glucagon. Front. Endocrinol. 2013, 4, 132. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Neely, B.A.; Soper, J.L.; Gulland, F.M.; Bell, P.D.; Kindy, M.; Arthur, J.M.; Janech, M.G. Proteomic analysis of cerebrospinal fluid in California sea lions (Zalophus californianus) with domoic acid toxicosis identifies proteins associated with neurodegeneration. Proteomics 2015, 15, 4051–4063. [Google Scholar] [CrossRef]
- Ordonez, Y.N.; Anton, R.F.; Davis, W.C. Quantification of total serum transferrin and transferrin sialoforms in human serum; an alternative method for the determination of carbohydrate-deficient transferrin in clinical samples. Anal. Methods 2014, 6, 3967–3974. [Google Scholar] [CrossRef]
- Johnson, S.P.; Venn-Watson, S.K.; Cassle, S.E.; Smith, C.R.; Jensen, E.D.; Ridgway, S.H. Use of phlebotomy treatment in Atlantic bottlenose dolphins with iron overload. J. Am. Vet. Med. Assoc. 2009, 235, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Leon, I.R.; Schwammle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef]
- Bryk, A.H.; Wisniewski, J.R. Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef]
- Martens, L.; Vizcaino, J.A. A Golden Age for Working with Public Proteomics Data. Trends Biochem. Sci. 2017, 42, 333–341. [Google Scholar] [CrossRef]
- Jagtap, P.D.; Johnson, J.E.; Onsongo, G.; Sadler, F.W.; Murray, K.; Wang, Y.; Shenykman, G.M.; Bandhakavi, S.; Smith, L.M.; Griffin, T.J. Flexible and accessible workflows for improved proteogenomic analysis using the Galaxy framework. J. Proteome Res. 2014, 13, 5898–5908. [Google Scholar] [CrossRef]
- Sheynkman, G.M.; Johnson, J.E.; Jagtap, P.D.; Shortreed, M.R.; Onsongo, G.; Frey, B.L.; Griffin, T.J.; Smith, L.M. Using Galaxy-P to leverage RNA-Seq for the discovery of novel protein variations. BMC Genom. 2014, 15, 703. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hs 34 | Hs 105 | Hs 108 | |
|---|---|---|---|
| Release date | Feb 2004 | Jun 2013 | Mar 2016 |
| Assembly name | NCBI 24 | GRCh37.p13 | GRCh38.p7 |
| Scaffold N50 | 29.1 Mbp | 45.0 Mbp | 59.4 Mbp |
| Coding sequences | 27,180 | 45,107 | 109,018 |
| Protein groups identified | 9762 | 10,059 | 10,219 |
| Proteins identified | 13,341 | 22,906 | 48,019 |
| Peptide groups identified | 175,895 | 184,580 | 184,806 |
| Peptide spectral matches | 390,909 | 405,852 | 405,950 |
| NP10 | 100.17 kDa | 101.87 kDa | 133.55 kDa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neely, B.A.; Ellisor, D.L.; Davis, W.C. Proteomics as a Metrological Tool to Evaluate Genome Annotation Accuracy Following De Novo Genome Assembly: A Case Study Using the Atlantic Bottlenose Dolphin (Tursiops truncatus). Genes 2023, 14, 1696. https://doi.org/10.3390/genes14091696
Neely BA, Ellisor DL, Davis WC. Proteomics as a Metrological Tool to Evaluate Genome Annotation Accuracy Following De Novo Genome Assembly: A Case Study Using the Atlantic Bottlenose Dolphin (Tursiops truncatus). Genes. 2023; 14(9):1696. https://doi.org/10.3390/genes14091696
Chicago/Turabian StyleNeely, Benjamin A., Debra L. Ellisor, and W. Clay Davis. 2023. "Proteomics as a Metrological Tool to Evaluate Genome Annotation Accuracy Following De Novo Genome Assembly: A Case Study Using the Atlantic Bottlenose Dolphin (Tursiops truncatus)" Genes 14, no. 9: 1696. https://doi.org/10.3390/genes14091696
APA StyleNeely, B. A., Ellisor, D. L., & Davis, W. C. (2023). Proteomics as a Metrological Tool to Evaluate Genome Annotation Accuracy Following De Novo Genome Assembly: A Case Study Using the Atlantic Bottlenose Dolphin (Tursiops truncatus). Genes, 14(9), 1696. https://doi.org/10.3390/genes14091696