Simultaneous Occurrence of Multiple Neoplasms in Children with Cancer Predisposition Syndromes: Collaborating with Abnormal Genes

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Molecular Analysis
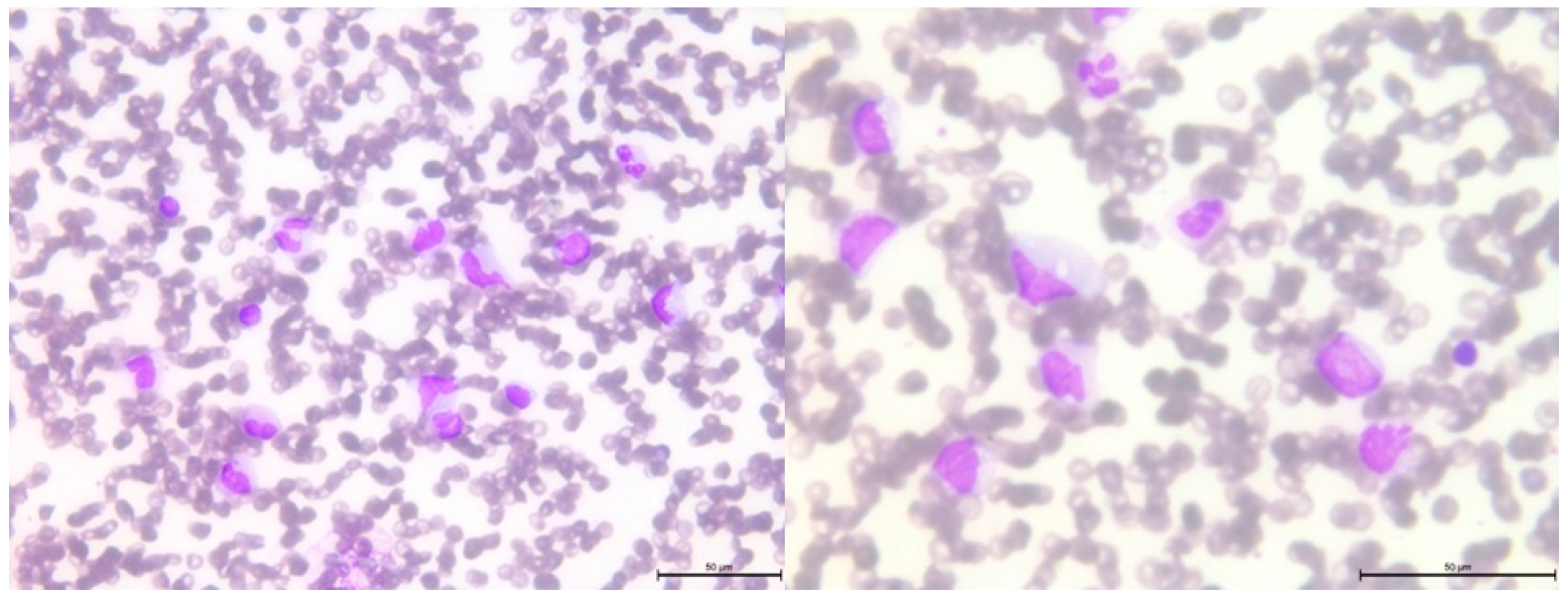
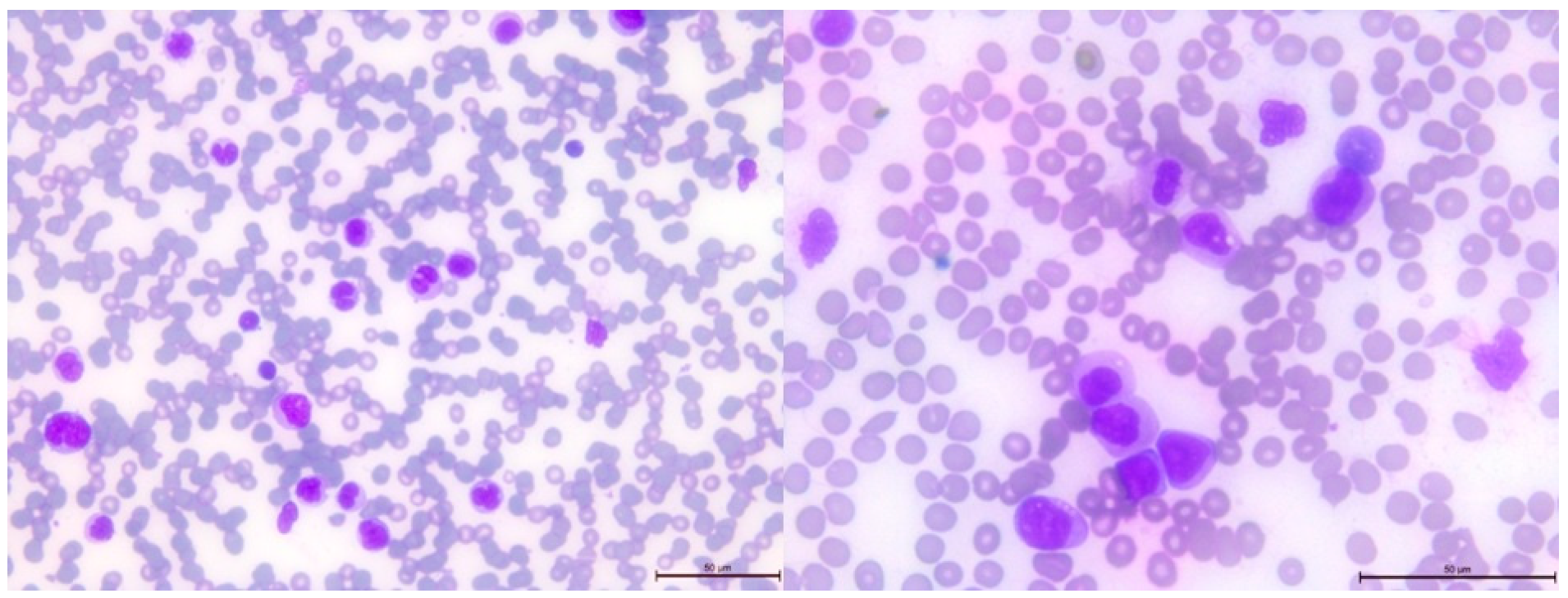
2.3. Cytological and Histological Analysis
2.4. Data Visualization
3. Results
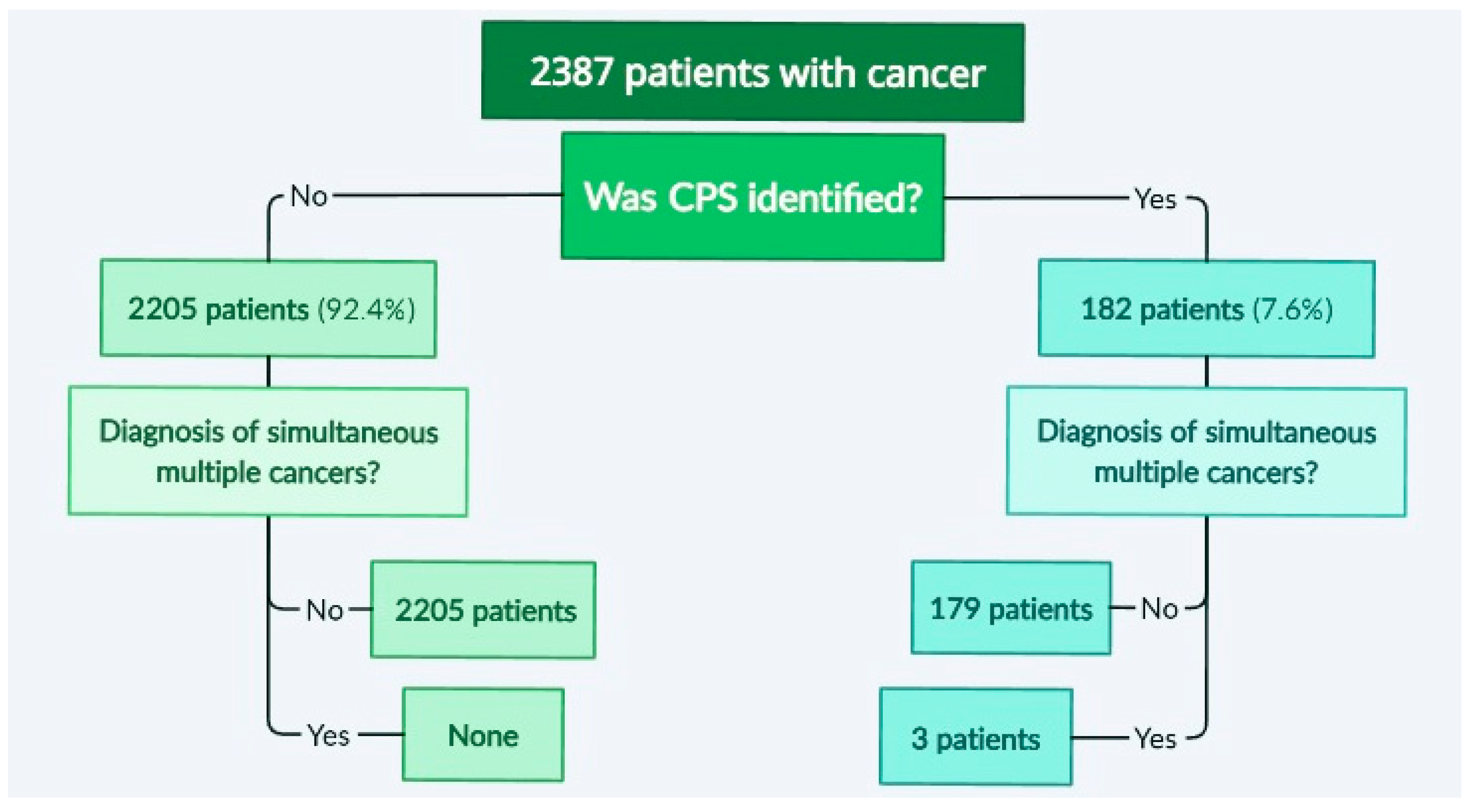
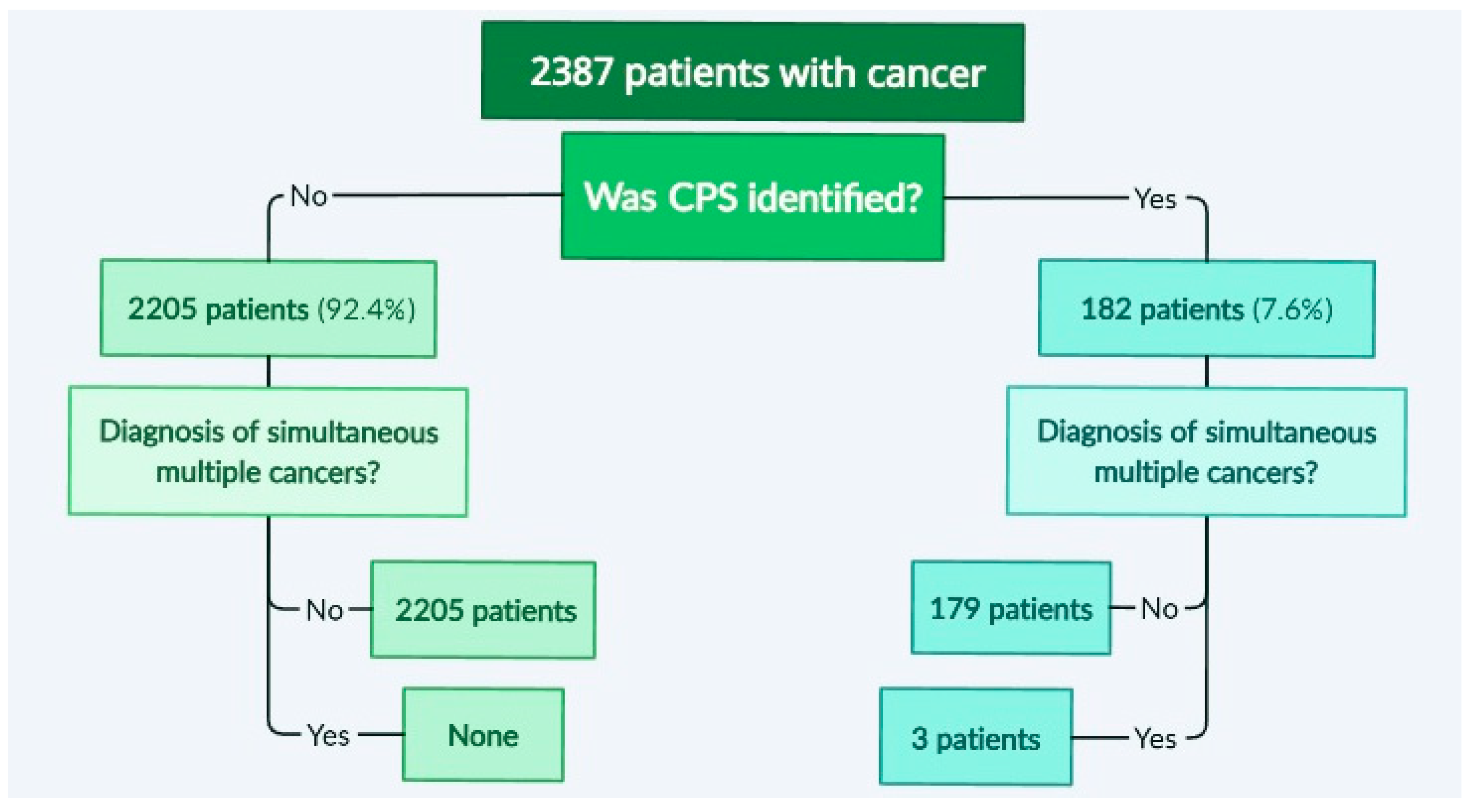
3.1. Identification of Cases with Simultaneous/Metachronous Multiple Cancers in a Studied Cohort
3.2. Pathogenic Variants of the BRCA2 Gene—Case 1
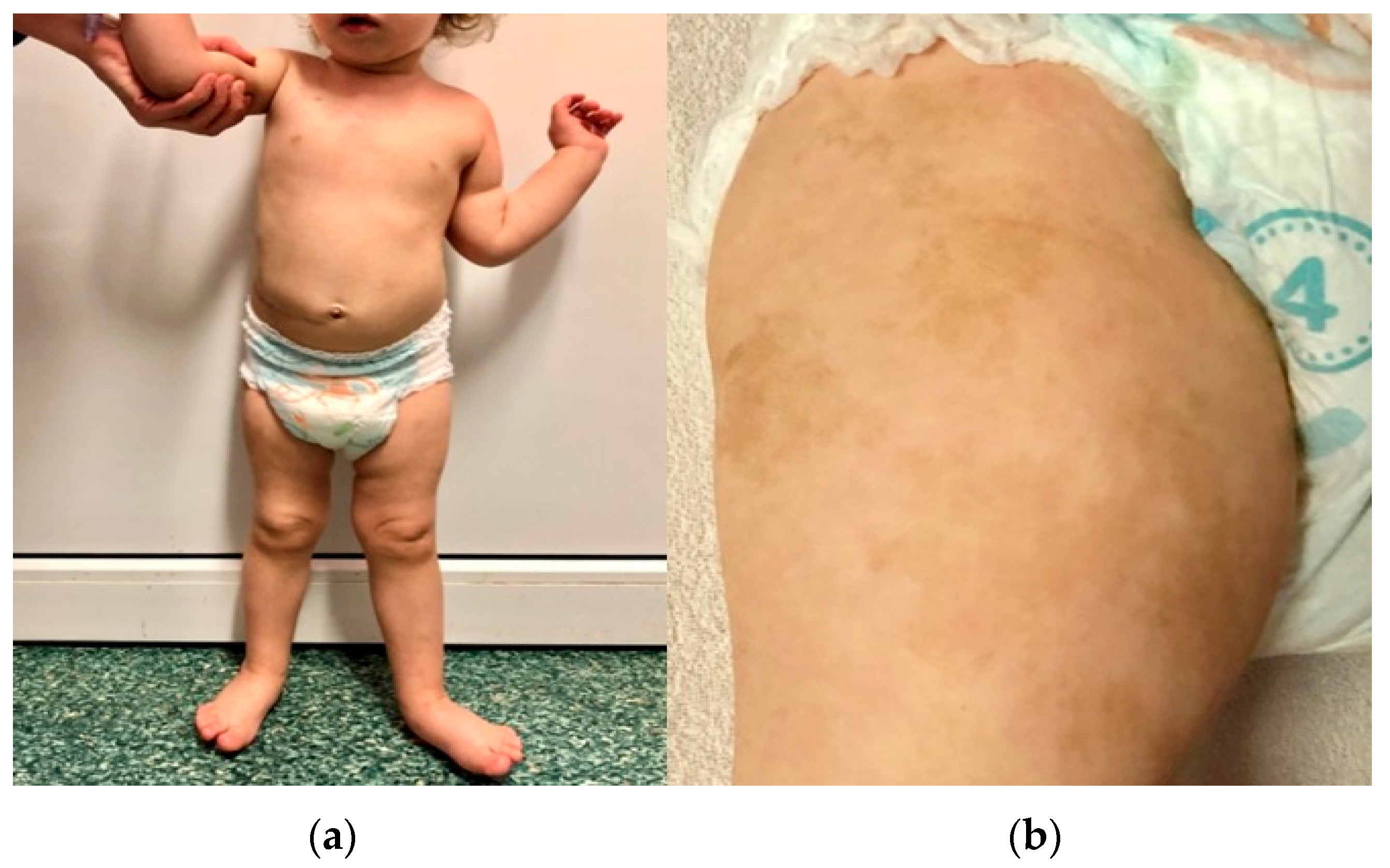
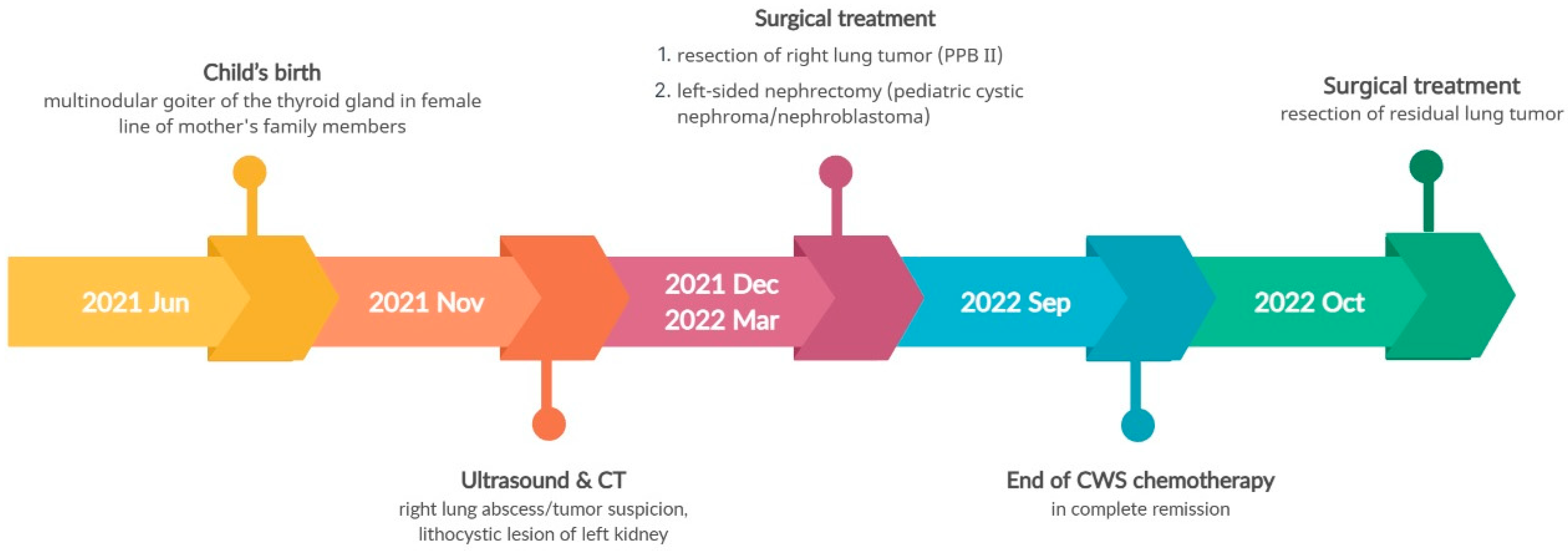
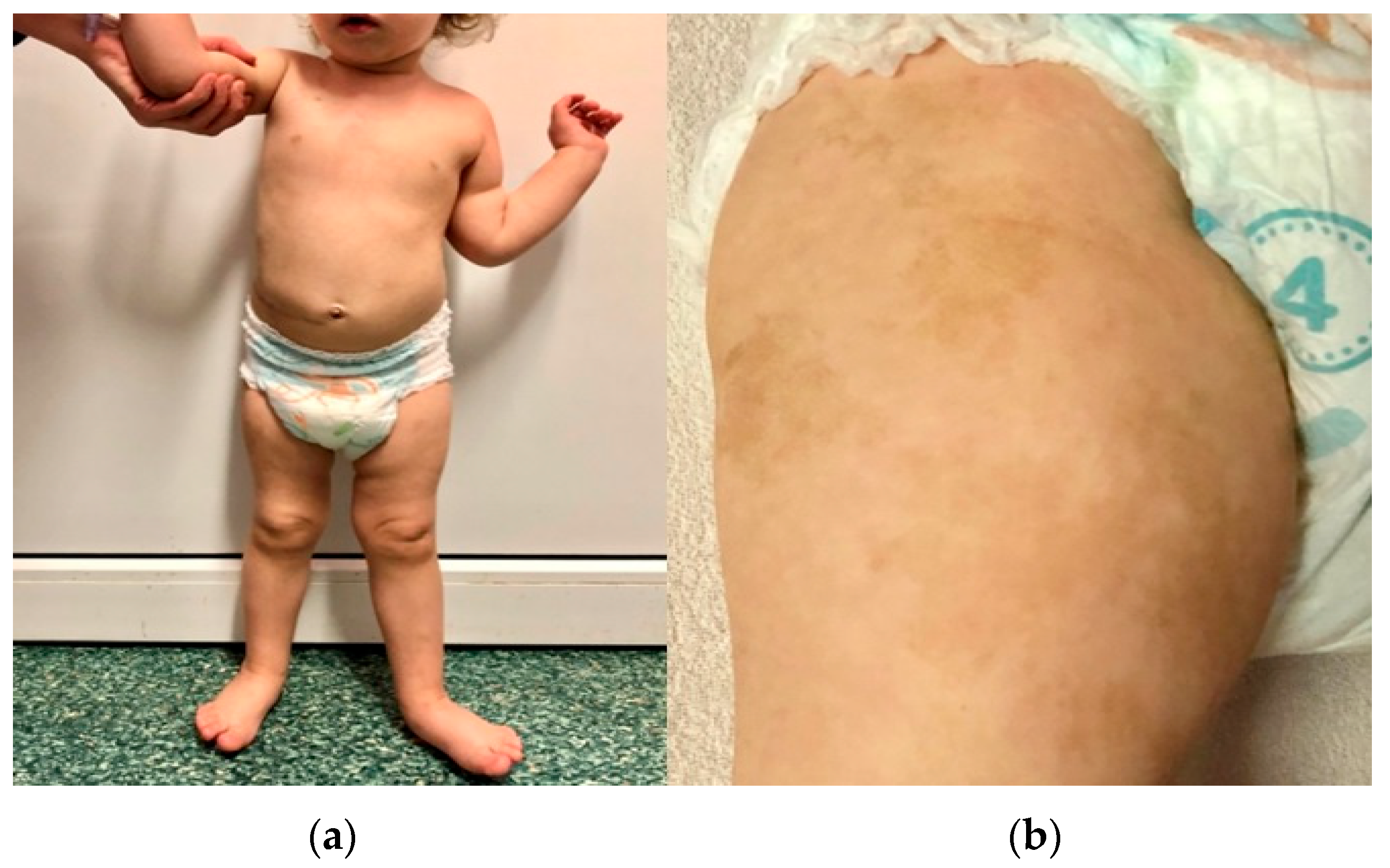
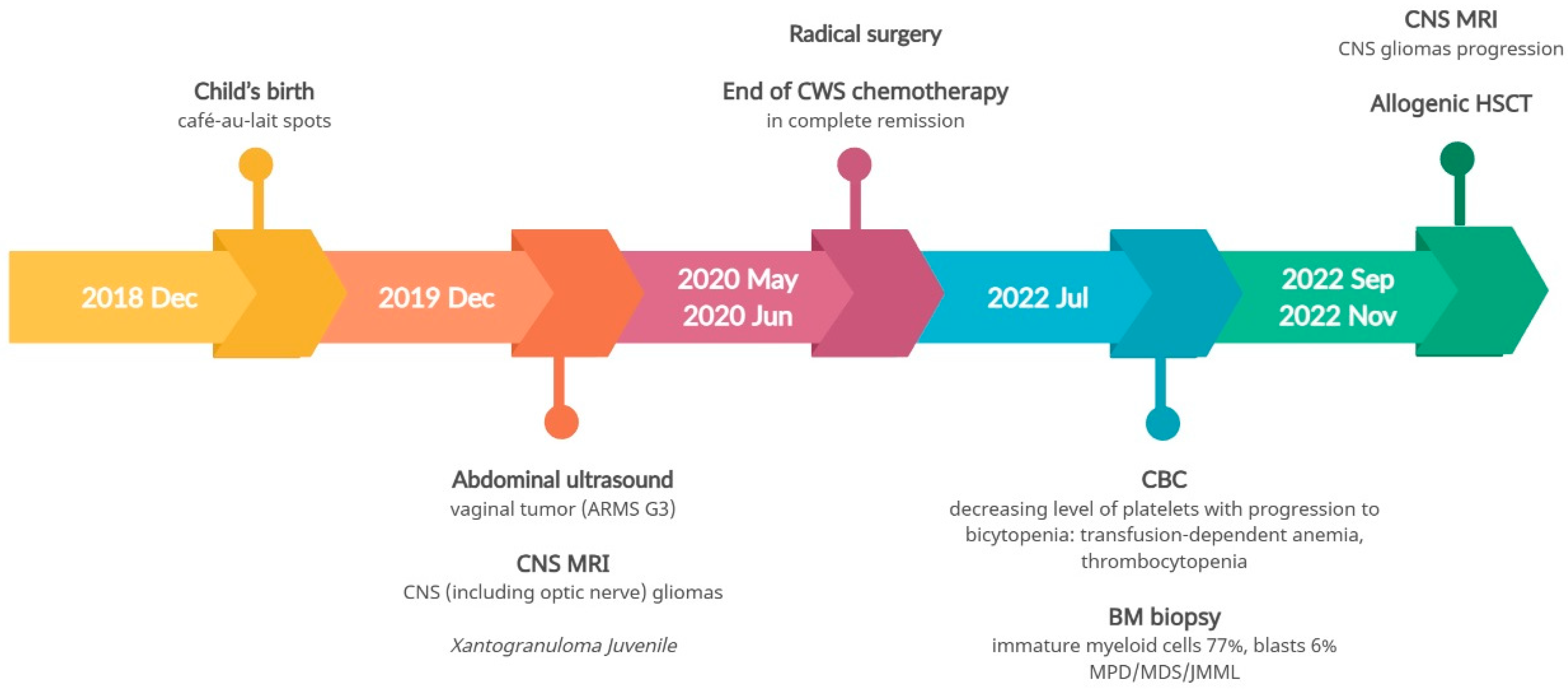
3.3. Pathogenic Variant of the NF1 Gene—Case 2
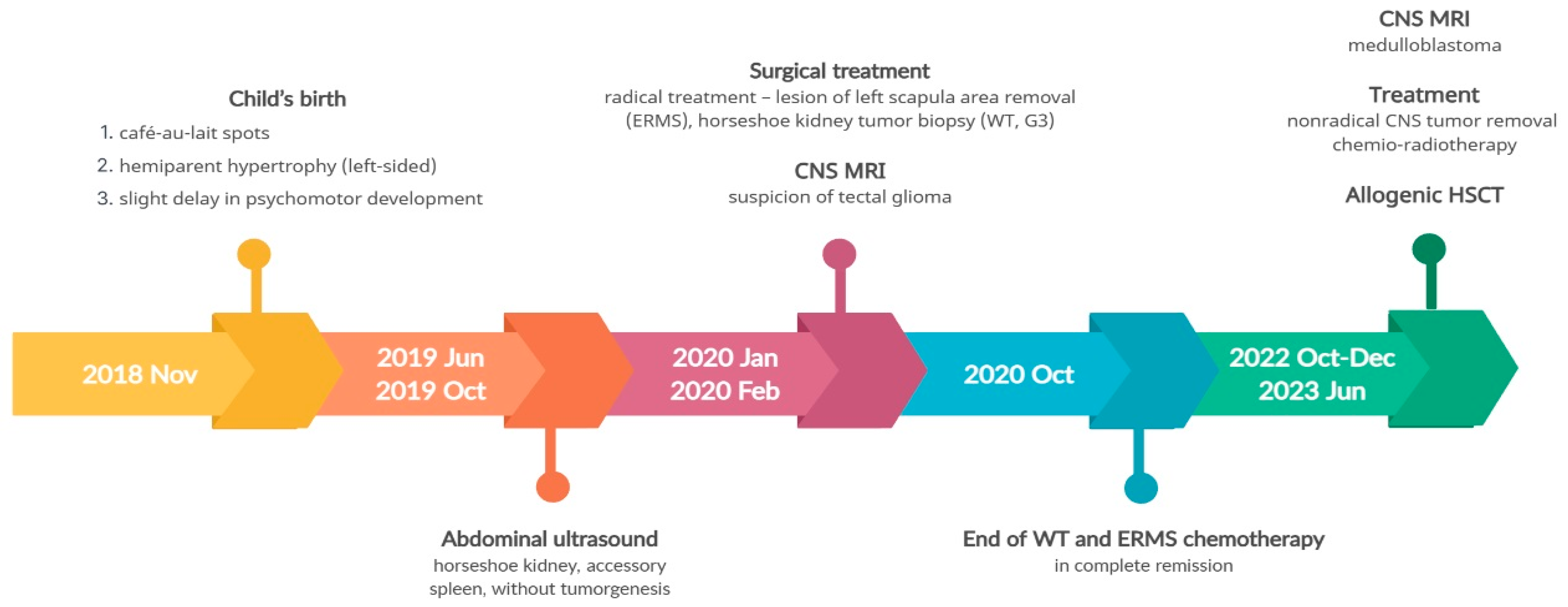
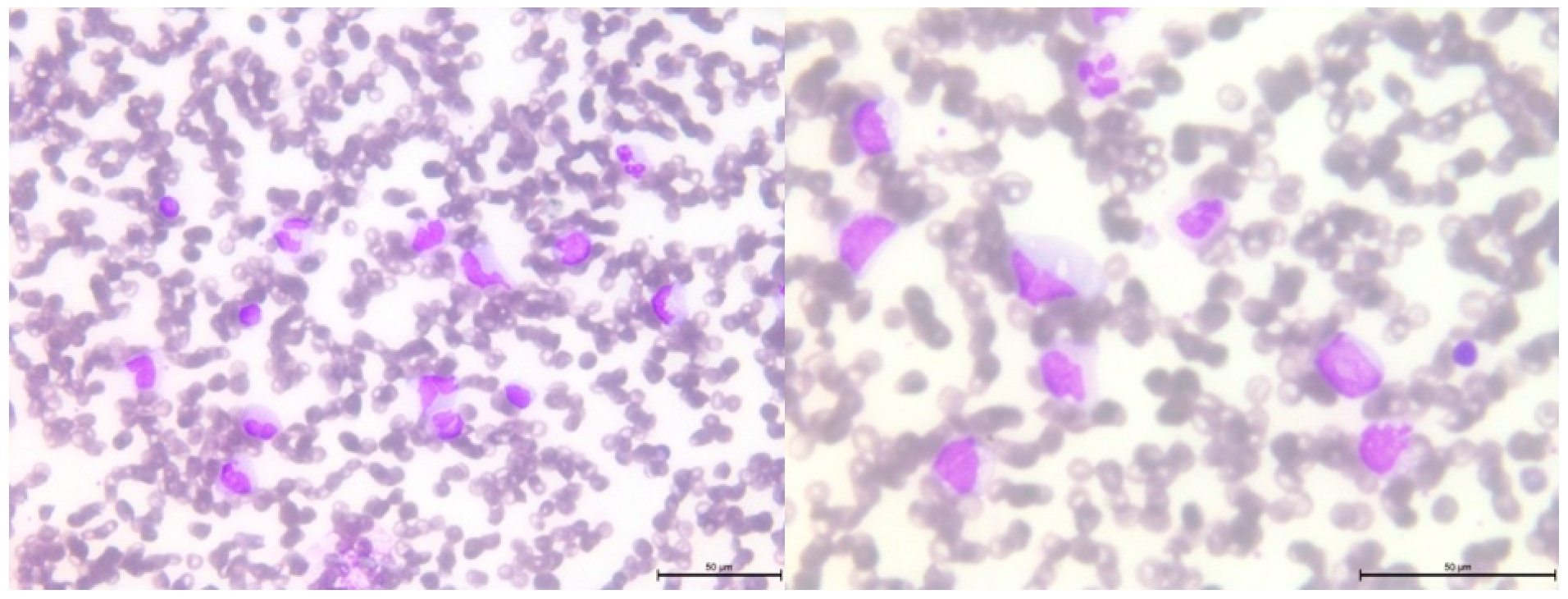
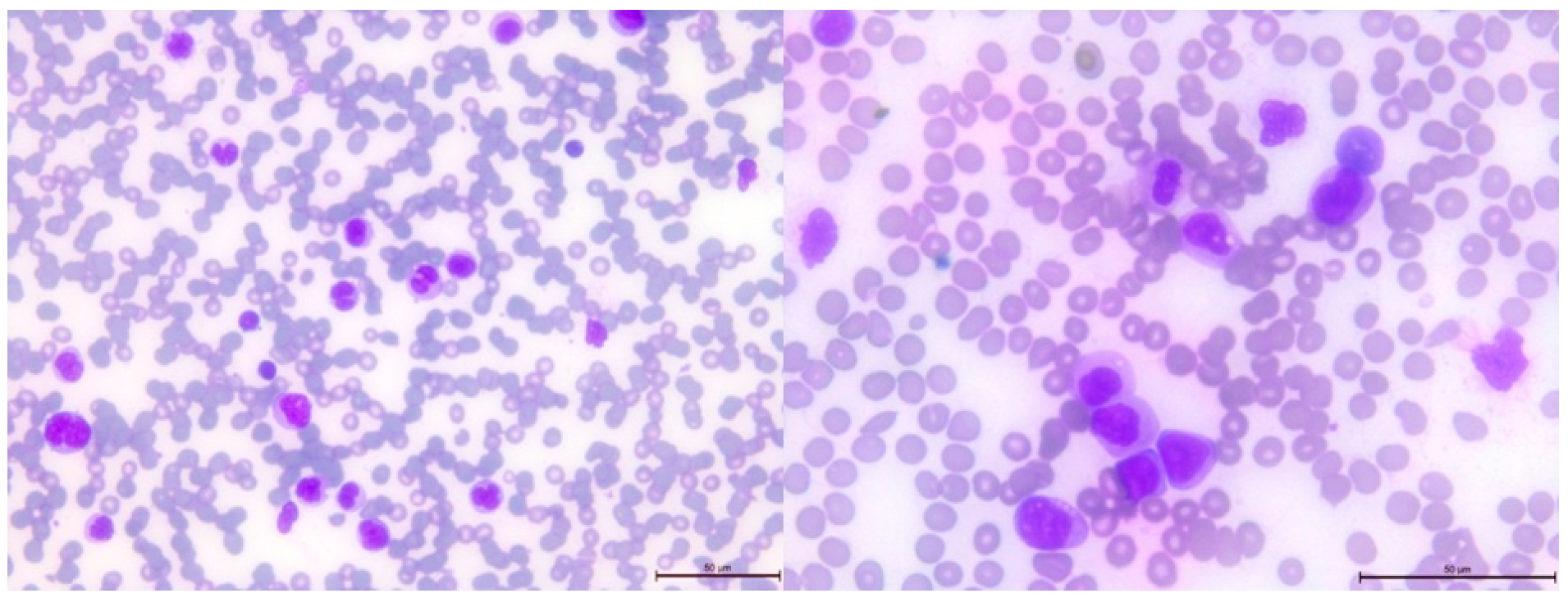
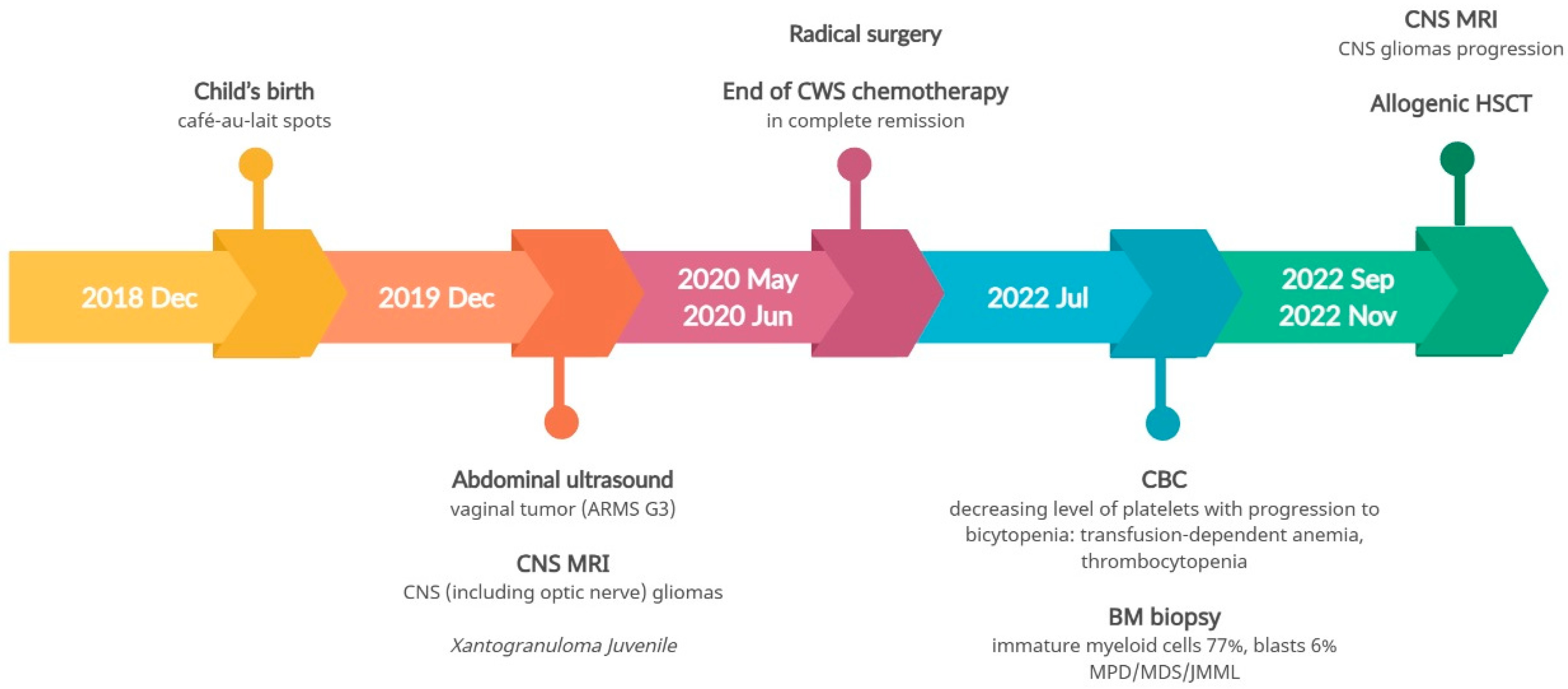
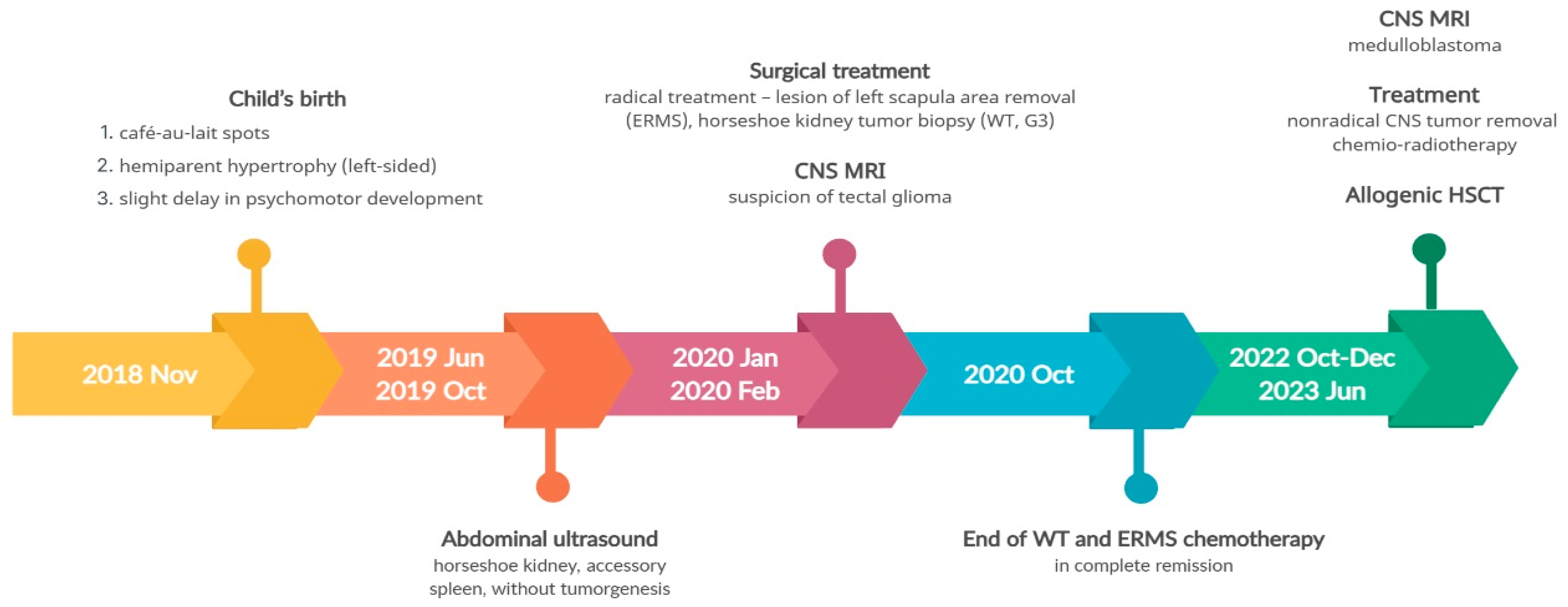
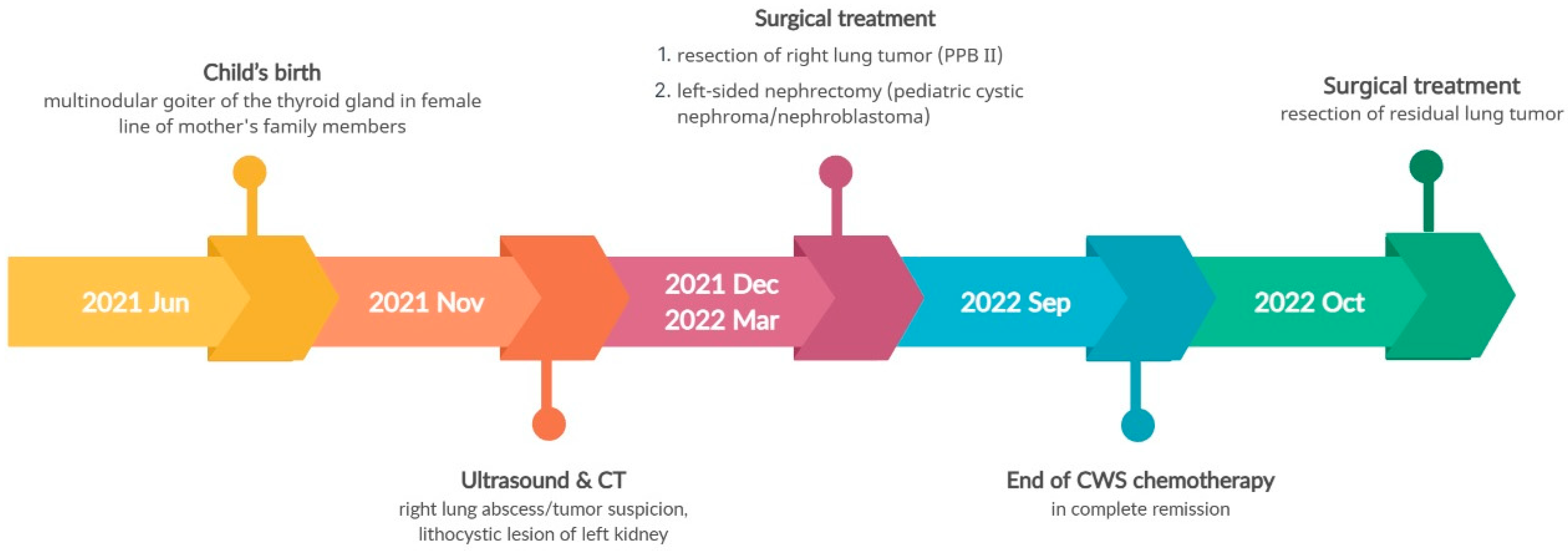
3.4. Pathogenic Variant of the DICER1 Gene—Case 3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A. Why do young people get cancer? Pediatr. Blood Cancer 2020, 67, e28335. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Yeh, J.M.; Bhakta, N.; Frazier, A.L.; Atun, R. Estimating the total incidence of global childhood cancer: A simulation-based analysis. Lancet Oncol. 2019, 20, 483–493. [Google Scholar] [CrossRef]
- Shahani, S.A.; Marcotte, E.L. Landscape of germline cancer predisposition mutations testing and management in pediatrics: Implications for research and clinical care. Front. Pediatr. 2022, 10, 1011873. [Google Scholar] [CrossRef]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discov. 2021, 11, 3008–3027. [Google Scholar] [CrossRef]
- Kratz, C.P.; Jongmans, M.C.; Cavé, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc. Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Benton, M.L.; Abraham, A.; LaBella, A.L.; Abbot, P.; Rokas, A.; Capra, J.A. The influence of evolutionary history on human health and disease. Nat. Rev. Genet. 2021, 22, 269–283. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Fair, D.; Potter, S.L.; Venkatramani, R. Challenges and solutions to the study of rare childhood tumors. Curr. Opin. Pediatr. 2020, 32, 7–12. [Google Scholar] [CrossRef]
- Scollon, S.; Anglin, A.K.; Thomas, M.; Turner, J.T.; Wolfe Schneider, K. A Comprehensive Review of Pediatric Tumors and Associated Cancer Predisposition Syndromes. J. Genet. Couns. 2017, 26, 387–434. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Nichols, K.E.; Schiffman, J.D.; Plon, S.E.; Brodeur, G.M. The Future of Surveillance in the Context of Cancer Predisposition: Through the Murky Looking Glass. Clin. Cancer Res. 2017, 23, e133–e137. [Google Scholar] [CrossRef] [PubMed]
- Druker, H.; Zelley, K.; McGee, R.B.; Scollon, S.R.; Kohlmann, W.K.; Schneider, K.A.; Wolfe Schneider, K. Genetic Counselor Recommendations for Cancer Predisposition Evaluation and Surveillance in the Pediatric Oncology Patient. Clin. Cancer Res. 2017, 23, e91–e97. [Google Scholar] [CrossRef] [PubMed]
- Krueger, H.; McLean, D.; Williams, D. The Prevention of Second Primary Cancers: A Resource for Clinicians and Health Managers; S. Karger AG: Basel, Switzerland, 2008. [Google Scholar]
- Zahnreich, S.; Schmidberger, H. Childhood Cancer: Occurrence, Treatment and Risk of Second Primary Malignancies. Cancers 2021, 13, 2607. [Google Scholar] [CrossRef] [PubMed]
- Bakhuizen, J.J.; Hopman, S.M.J.; Bosscha, M.I.; Dommering, C.J.; van den Heuvel-Eibrink, M.M.; Hol, J.A.; Kester, L.A.; Koudijs, M.J.; Langenberg, K.P.S.; Loeffen, J.L.C.; et al. Assessment of Cancer Predisposition Syndromes in a National Cohort of Children with a Neoplasm. JAMA Netw. Open 2023, 6, e2254157. [Google Scholar] [CrossRef]
- Duzkale, N.; Kandemir, O. The Relationship of Mutation Carriage of BRCA1/2 and Family History in Triple-Negative Breast Cancer: Experience from a Diagnostic Center in Turkey. Eur. J. Breast Health 2021, 17, 137–144. [Google Scholar] [CrossRef]
- Offit, K.; Levran, O.; Mullaney, B.; Mah, K.; Nafa, K.; Batish, S.D.; Diotti, R.; Schneider, H.; Deffenbaugh, A.; Scholl, T.; et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J. Natl. Cancer Inst. 2003, 95, 1548–1551. [Google Scholar] [CrossRef]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Hata, A.; Kashima, R. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 121–134. [Google Scholar] [CrossRef]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J. Med. Genet. 2014, 51, 71–75. [Google Scholar] [CrossRef]
- Rosenbaum, T.; Wimmer, K. Neurofibromatosis type 1 (NF1) and associated tumors. Klin. Pädiatrie 2014, 226, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.C.; Jorcyk, C.L.; Oxford, J.T. DICER1 syndrome: DICER1 mutations in rare cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- González, I.A.; Stewart, D.R.; Schultz, K.A.P.; Field, A.P.; Hill, D.A.; Dehner, L.P. DICER1 tumor predisposition syndrome: An evolving story initiated with the pleuropulmonary blastoma. Mod. Pathol. 2022, 35, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.N.; Rhee, D.; Tracy, E.T.; Aldrink, J.H.; Baertschiger, R.M.; Lautz, T.B.; Glick, R.D.; Rodeberg, D.A.; Ehrlich, P.F.; Christison-Lagay, E. Pediatric solid tumors and associated cancer predisposition syndromes: Workup, management, and surveillance. A summary from the APSA Cancer Committee. J. Pediatr. Surg. 2022, 57, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.; Pencheva, B.B.; Patil, V.M.; Fangusaro, J. Nervous system (NS) Tumors in Cancer Predisposition Syndromes. Neurotherapeutics 2022, 19, 1752–1771. [Google Scholar] [CrossRef]
- Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes 2021, 12, 1520. [Google Scholar] [CrossRef]
- Radtke, H.B.; Berger, A.; Skelton, T.; Goetsch Weisman, A. Neurofibromatosis Type 1 (NF1): Addressing the Transition from Pediatric to Adult Care. Pediatr. Health Med. Ther. 2023, 14, 19–32. [Google Scholar] [CrossRef]
- Caroleo, A.M.; De Ioris, M.A.; Boccuto, L.; Alessi, I.; Del Baldo, G.; Cacchione, A.; Agolini, E.; Rinelli, M.; Serra, A.; Carai, A.; et al. DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front. Oncol. 2020, 10, 614541. [Google Scholar] [CrossRef]
- Ferdynus, M.P. Why the term ‘persistent therapy’ is not worse than the term ‘medical futility’. J. Med. Ethics 2022, 48, 350–352. [Google Scholar] [CrossRef]
- Rossini, L.; Durante, C.; Bresolin, S.; Opocher, E.; Marzollo, A.; Biffi, A. Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy. Cancers 2022, 14, 3741. [Google Scholar] [CrossRef]
- Al-Sarhani, H.; Gottumukkala, R.V.; Grasparil, A.D.S., 2nd; Tung, E.L.; Gee, M.S.; Greer, M.C. Screening of cancer predisposition syndromes. Pediatr. Radiol. 2022, 52, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Schimmel, M.; Dunstheimer, D.; Nemes, K.; Richter, M.; Streble, J.; Vollert, K.; Walden, U.; Frühwald, M.C.; Kuhlen, M. The need for tumor surveillance of children and adolescents with cancer predisposition syndromes: A retrospective cohort study in a tertiary-care children’s hospital. Eur. J. Pediatr. 2022, 181, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Khanna, L.; Prasad, S.R.; Yedururi, S.; Parameswaran, A.M.; Marcal, L.P.; Sandrasegaran, K.; Tirumani, S.H.; Menias, C.O.; Katabathina, V.S. Second Malignancies after Radiation Therapy: Update on Pathogenesis and Cross-sectional Imaging Findings. Radiographics 2021, 41, 876–894. [Google Scholar] [CrossRef]
- Tanjak, P.; Suktitipat, B.; Vorasan, N.; Juengwiwattanakitti, P.; Thiengtrong, B.; Songjang, C.; Therasakvichya, S.; Laiteerapong, S.; Chinswangwatanakul, V. Risks and cancer associations of metachronous and synchronous multiple primary cancers: A 25-year retrospective study. BMC Cancer 2021, 21, 1045. [Google Scholar] [CrossRef]
- Bhatia, S.; Chen, Y.; Wong, F.L.; Hageman, L.; Smith, K.; Korf, B.; Cannon, A.; Leidy, D.J.; Paz, A.; Andress, J.E.; et al. Subsequent Neoplasms after a Primary Tumor in Individuals with Neurofibromatosis Type 1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3050–3058. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function | Mutation Consequences | Health Conditions Related to Genetic Changes |
|---|---|---|---|
| BRCA2 |
| Most pathogenic variants of the BRCA2 gene result in the production of an abnormally small and nonfunctional form of the BRCA2 protein from one copy of the gene in each cell. This leads to a reduced amount of available protein, which impairs efficient DNA repair. The accumulation of these defects can trigger uncontrolled cell growth and division, ultimately leading to carcinogenesis. |
|
| NF1 |
| Many pathogenic variants of the NF1 gene lead to the production of an extremely short version of neurofibromin, which is unable to inhibit cell division. The presence of these variants may result in a constitutive activation of the signal transduction pathway, as the G proteins are constantly active due to the failure to degrade the attached GTP molecule. The loss of protein activity also increases susceptibility to harmful UV effects. |
|
| DICER1 |
| DICER1 can be considered both a tumor suppressor gene, due to loss-of-function mutations, and an oncogene, due to gain-of-function mutations. It is believed to function as a haploinsufficient tumor suppressor gene, where the loss of one allele leads to tumor progression, but the loss of both alleles has an inhibitory effect on tumor development, implying that one intact allele is necessary for cell survival. |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telman, G.; Strauss, E.; Sosnowska-Sienkiewicz, P.; Halasz, M.; Januszkiewicz-Lewandowska, D. Simultaneous Occurrence of Multiple Neoplasms in Children with Cancer Predisposition Syndromes: Collaborating with Abnormal Genes. Genes 2023, 14, 1670. https://doi.org/10.3390/genes14091670
Telman G, Strauss E, Sosnowska-Sienkiewicz P, Halasz M, Januszkiewicz-Lewandowska D. Simultaneous Occurrence of Multiple Neoplasms in Children with Cancer Predisposition Syndromes: Collaborating with Abnormal Genes. Genes. 2023; 14(9):1670. https://doi.org/10.3390/genes14091670
Chicago/Turabian StyleTelman, Gabriela, Ewa Strauss, Patrycja Sosnowska-Sienkiewicz, Magdalena Halasz, and Danuta Januszkiewicz-Lewandowska. 2023. "Simultaneous Occurrence of Multiple Neoplasms in Children with Cancer Predisposition Syndromes: Collaborating with Abnormal Genes" Genes 14, no. 9: 1670. https://doi.org/10.3390/genes14091670
APA StyleTelman, G., Strauss, E., Sosnowska-Sienkiewicz, P., Halasz, M., & Januszkiewicz-Lewandowska, D. (2023). Simultaneous Occurrence of Multiple Neoplasms in Children with Cancer Predisposition Syndromes: Collaborating with Abnormal Genes. Genes, 14(9), 1670. https://doi.org/10.3390/genes14091670