
Genetic Ancestry Estimates within Dutch Family Units and Across Genotyping Arrays: Insights from Empirical Analysis Using Two Estimation Methods

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Selection and Genotyping
2.2. Dataset Curation
2.3. Final Sample Composition
2.4. Reference Dataset
2.5. Principal Components Analysis
2.6. ADMIXTURE Analysis
2.7. Statistical Analysis
3. Results
3.1. Principal Components Analysis
3.2. ADMIXTURE Analysis
3.3. Ancestry Outliers—PCA vs. ADMIXTURE
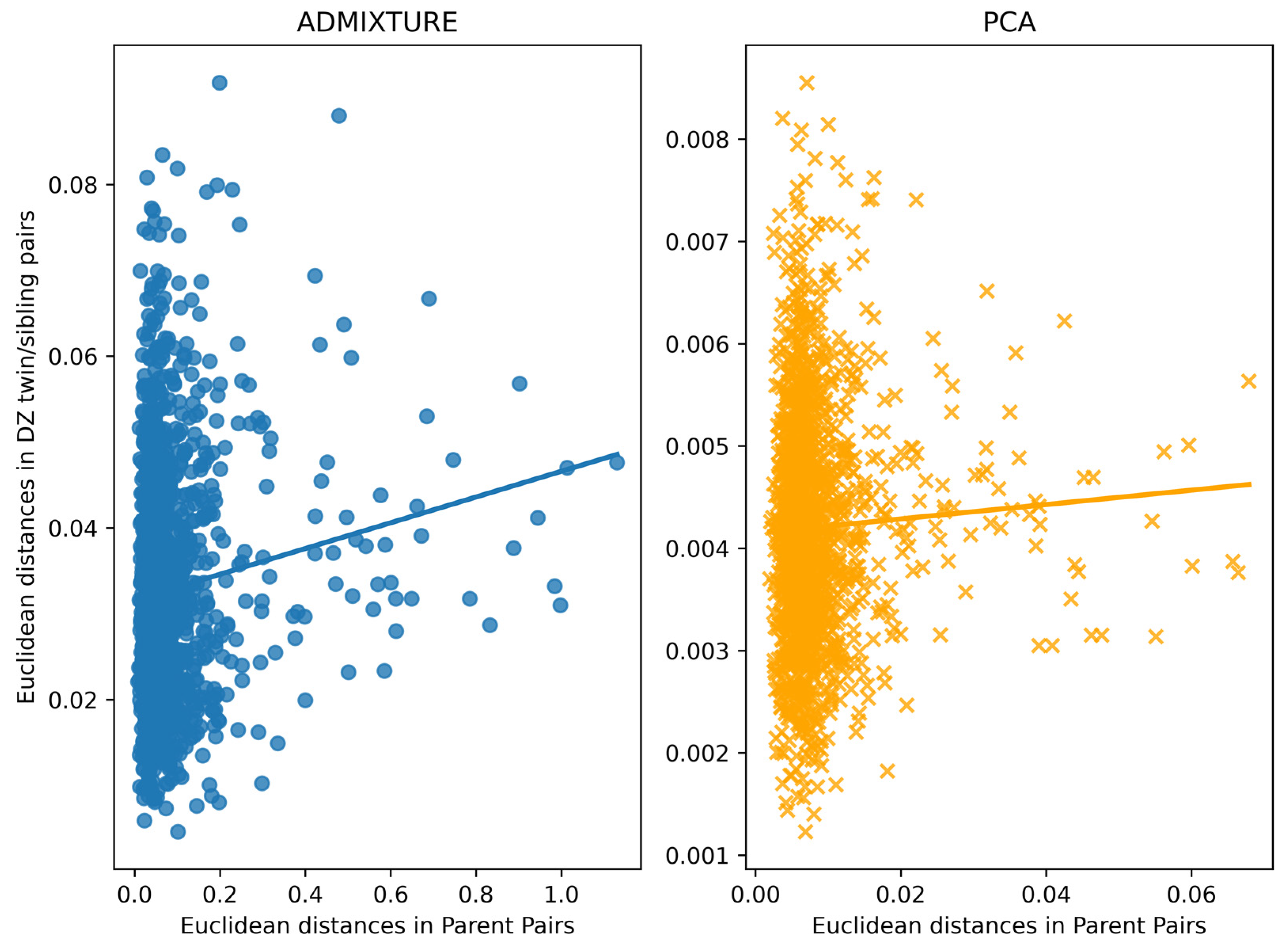
3.4. Assessment of Within-Family Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Code Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Novembre, J.; Stephens, M. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 2008, 40, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Novembre, J.; Johnson, T.; Bryc, K.; Kutalik, Z.; Boyko, A.R.; Auton, A.; Indap, A.; King, K.S.; Bergmann, S.; Nelson, M.R.; et al. Genes mirror geography within Europe. Nature 2008, 456, 98–101. [Google Scholar] [CrossRef]
- Reich, D.; Price, A.L.; Patterson, N. Principal component analysis of genetic data. Nat. Genet. 2008, 40, 491–492. [Google Scholar] [CrossRef]
- Abdellaoui, A.; Hottenga, J.-J.; de Knijff, P.; Nivard, M.G.; Xiao, X.; Scheet, P.; Brooks, A.; Ehli, E.A.; Hu, Y.; Davies, G.E.; et al. Population structure, migration, and diversifying selection in the Netherlands. Eur. J. Hum. Genet. 2013, 21, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Prive, F.; Luu, K.; Blum, M.G.B.; McGrath, J.J.; Vilhjalmsson, B.J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 2020, 36, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Weale, M.E.; Patterson, N.; Myers, S.R.; Need, A.C.; Shianna, K.V.; Ge, D.; Rotter, J.I.; Torres, E.; Taylor, K.D.; et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 2008, 83, 132–135. [Google Scholar] [CrossRef]
- Zou, F.; Lee, S.; Knowles, M.R.; Wright, F.A. Quantification of population structure using correlated SNPs by shrinkage principal components. Hum. Hered. 2010, 70, 9–22. [Google Scholar] [CrossRef]
- Prive, F.; Aschard, H.; Ziyatdinov, A.; Blum, M.G.B. Efficient analysis of large-scale genome-wide data with two R packages: Bigstatsr and bigsnpr. Bioinformatics 2018, 34, 2781–2787. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Peng, J.; Wang, P.; Risch, N.J. Estimation of individual admixture: Analytical and study design considerations. Genet. Epidemiol. 2005, 28, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Lawson, D.J.; van Dorp, L.; Falush, D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat. Commun. 2018, 9, 3258. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Engelhardt, B.E.; Stephens, M. Analysis of population structure: A unifying framework and novel methods based on sparse factor analysis. PLoS Genet. 2010, 6, e1001117. [Google Scholar] [CrossRef]
- McVean, G. A genealogical interpretation of principal components analysis. PLoS Genet. 2009, 5, e1000686. [Google Scholar] [CrossRef]
- Ma, J.; Amos, C.I. Principal components analysis of population admixture. PLoS ONE 2012, 7, e40115. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zheng, X.; Weir, B.S. Eigenanalysis of SNP data with an identity by descent interpretation. Popul. Biol. 2016, 107, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Cardon, L.R.; Cookson, W.O. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 2000, 66, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, B.; Visscher, P.M.; McRae, A.F. Family-based genome-wide association studies. Pharmacogenomics 2009, 10, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Brumpton, B.; Sanderson, E.; Heilbron, K.; Hartwig, F.P.; Harrison, S.; Vie, G.A.; Cho, Y.; Howe, L.D.; Hughes, A.; Boomsma, D.I.; et al. Avoiding dynastic, assortative mating, and population stratification biases in Mendelian randomization through within-family analyses. Nat. Commun. 2020, 11, 3519. [Google Scholar] [CrossRef]
- Howe, L.J.; Nivard, M.G.; Morris, T.T.; Hansen, A.F.; Rasheed, H.; Cho, Y.; Chittoor, G.; Ahlskog, R.; Lind, P.A.; Palviainen, T.; et al. Within-sibship genome-wide association analyses decrease bias in estimates of direct genetic effects. Nat. Genet. 2022, 54, 581–592. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef]
- Genome of the Netherlands, C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef]
- Willemsen, G.; Vink, J.M.; Abdellaoui, A.; den Braber, A.; van Beek, J.H.; Draisma, H.H.; van Dongen, J.; van ’t Ent, D.; Geels, L.M.; van Lien, R.; et al. The Adult Netherlands Twin Register: Twenty-five years of survey and biological data collection. Twin Res. Hum. Genet. 2013, 16, 271–281. [Google Scholar] [CrossRef]
- van Beijsterveldt, C.E.; Groen-Blokhuis, M.; Hottenga, J.J.; Franic, S.; Hudziak, J.J.; Lamb, D.; Huppertz, C.; de Zeeuw, E.; Nivard, M.; Schutte, N.; et al. The Young Netherlands Twin Register (YNTR): Longitudinal twin and family studies in over 70,000 children. Twin Res. Hum. Genet. 2013, 16, 252–267. [Google Scholar] [CrossRef]
- Min, J.L.; Lakenberg, N.; Bakker-Verweij, M.; Suchiman, E.; Boomsma, D.I.; Slagboom, P.E.; Meulenbelt, I. High microsatellite and SNP genotyping success rates established in a large number of genomic DNA samples extracted from mouth swabs and genotypes. Twin Res. Hum. Genet. 2006, 9, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Ehli, E.A.; Abdellaoui, A.; Fedko, I.O.; Grieser, C.; Nohzadeh-Malakshah, S.; Willemsen, G.; de Geus, E.J.; Boomsma, D.I.; Davies, G.E.; Hottenga, J.J. A method to customize population-specific arrays for genome-wide association testing. Eur. J. Hum. Genet. 2017, 25, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.J.; Hottenga, J.J.; Mbarek, H.; Finnicum, C.T.; Ehli, E.A.; Hur, Y.M.; Martin, N.G.; de Geus, E.J.C.; Boomsma, D.I.; Davies, G.E. Genetic Similarity Assessment of Twin-Family Populations by Custom-Designed Genotyping Array. Twin Res. Hum. Genet. 2019, 22, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
- Jonsson, H.; Magnusdottir, E.; Eggertsson, H.P.; Stefansson, O.A.; Arnadottir, G.A.; Eiriksson, O.; Zink, F.; Helgason, E.A.; Jonsdottir, I.; Gylfason, A.; et al. Differences between germline genomes of monozygotic twins. Nat. Genet. 2021, 53, 27–34. [Google Scholar] [CrossRef]
- Ouwens, K.G.; Jansen, R.; Tolhuis, B.; Slagboom, P.E.; Penninx, B.; Boomsma, D.I. A characterization of postzygotic mutations identified in monozygotic twins. Hum. Mutat. 2018, 39, 1393–1401. [Google Scholar] [CrossRef]
- Royal, C.D.; Novembre, J.; Fullerton, S.M.; Goldstein, D.B.; Long, J.C.; Bamshad, M.J.; Clark, A.G. Inferring genetic ancestry: Opportunities, challenges, and implications. Am. J. Hum. Genet. 2010, 86, 661–673. [Google Scholar] [CrossRef]
- Akey, J.M.; Eberle, M.A.; Rieder, M.J.; Carlson, C.S.; Shriver, M.D.; Nickerson, D.A.; Kruglyak, L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004, 2, e286. [Google Scholar] [CrossRef]
- Lemke, A.A.; Esplin, E.D.; Goldenberg, A.J.; Gonzaga-Jauregui, C.; Hanchard, N.A.; Harris-Wai, J.; Ideozu, J.E.; Isasi, R.; Landstrom, A.P.; Prince, A.E. Addressing underrepresentation in genomics research through community engagement. Am. J. Hum. Genet. 2022, 109, 1563–1571. [Google Scholar] [CrossRef]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 1080. [Google Scholar] [CrossRef]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef]
- Kang, S.J.; Larkin, E.K.; Song, Y.; Barnholtz-Sloan, J.; Baechle, D.; Feng, T.; Zhu, X. Assessing the impact of global versus local ancestry in association studies. BMC Proc. 2009, 3 (Suppl. 7), S107. [Google Scholar] [CrossRef]
- Feng, Q.; Abraham, J.; Feng, T.; Song, Y.; Elston, R.C.; Zhu, X. A method to correct for population structure using a segregation model. BMC Proc. 2009, 3 (Suppl. 7), S104. [Google Scholar] [CrossRef]
- Thornton, T.; Conomos, M.P.; Sverdlov, S.; Blue, E.M.; Cheung, C.Y.; Glazner, C.G.; Lewis, S.M.; Wijsman, E.M. Estimating and adjusting for ancestry admixture in statistical methods for relatedness inference, heritability estimation, and association testing. BMC Proc. 2014, 8, S5. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. Principal Component Analyses (PCA)-based findings in population genetic studies are highly biased and must be reevaluated. Sci. Rep. 2022, 12, 14683. [Google Scholar] [CrossRef] [PubMed]
- Akond, Z.; Ahsan, M.A.; Alam, M.; Mollah, M.N.H. Robustification of GWAS to explore effective SNPs addressing the challenges of hidden population stratification and polygenic effects. Sci. Rep. 2021, 11, 13060. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, S.; Cooper, R.S.; Elston, R.C. A unified association analysis approach for family and unrelated samples correcting for stratification. Am. J. Hum. Genet. 2008, 82, 352–365. [Google Scholar] [CrossRef]
- Conomos, M.P.; Miller, M.B.; Thornton, T.A. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol. 2015, 39, 276–293. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotyping Platform | Unique Families | Unique Individuals | MZ Twin Pairs | DZ Twin/Sibling Pairs | Parent–Offspring Pairs | Parent Pairs |
|---|---|---|---|---|---|---|
| AFFY6 | 2800 | 7575 | 1279 | 2966 | 2849 | 438 |
| AXIOM | 734 | 2593 | 433 | 591 | 2222 | 448 |
| ILLGSA | 3562 | 11,597 | 1546 | 3689 | 8366 | 1805 |
| Across all platforms | 6361 | 21,177 | 3528 | 7246 | 13,437 | 2691 |
| Harmonized | 6692 | 23,086 | 3406 | 8464 | 16,878 | 3023 |
| Min. and Max. Thresholds Determined by GoNL 1 | Min. and Max. Thresholds Determined by CEU 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PCA 3 | ADMIXTURE 4 | Common Outliers 5 | PCA 3 | ADMIXTURE 4 | Common Outliers 5 | |||||
| Dataset (N) | Outliers (%) | Inliers (%) | Outliers (%) | Inliers (%) | Outliers (%) | Inliers (%) | Outliers (%) | Inliers (%) | ||
| AFFY6 (8744) | 525 (6.0%) | 8219 (94.0%) | 695 (7.9%) | 8049 (92.1%) | 457 | 818 (9.4%) | 7926 (90.6%) | 2419 (27.7%) | 6325 (72.3%) | 704 |
| AXIOM (3361) | 249 (7.4%) | 3112 (92.6%) | 341 (10.1%) | 3020 (89.9%) | 224 | 451 (13.4%) | 2910 (86.6%) | 896 (26.7%) | 2465 (73.3%) | 382 |
| ILLGSA (13,686) | 933 (6.8%) | 12,753 (93.2%) | 954 (7.0%) | 12,732 (93.0%) | 809 | 1169 (8.5%) | 12,517 (91.5%) | 2501 (18.3%) | 11,185 (81.7%) | 891 |
| Harmonized (25,005) | 1794 (7.2%) | 23,211 (92.8%) | 5617 (22.5%) | 19,388 (77.5%) | 1584 | 3438 (13.7%) | 21,567 (86.3%) | 12,501 (50.0%) | 12,504 (50.0%) | 2796 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, J.J.; Ahmed, T.; Finnicum, C.T.; Zwinderman, K.; Ehli, E.A.; Boomsma, D.I.; Hottenga, J.J. Genetic Ancestry Estimates within Dutch Family Units and Across Genotyping Arrays: Insights from Empirical Analysis Using Two Estimation Methods. Genes 2023, 14, 1497. https://doi.org/10.3390/genes14071497
Beck JJ, Ahmed T, Finnicum CT, Zwinderman K, Ehli EA, Boomsma DI, Hottenga JJ. Genetic Ancestry Estimates within Dutch Family Units and Across Genotyping Arrays: Insights from Empirical Analysis Using Two Estimation Methods. Genes. 2023; 14(7):1497. https://doi.org/10.3390/genes14071497
Chicago/Turabian StyleBeck, Jeffrey J., Talitha Ahmed, Casey T. Finnicum, Koos Zwinderman, Erik A. Ehli, Dorret I. Boomsma, and Jouke Jan Hottenga. 2023. "Genetic Ancestry Estimates within Dutch Family Units and Across Genotyping Arrays: Insights from Empirical Analysis Using Two Estimation Methods" Genes 14, no. 7: 1497. https://doi.org/10.3390/genes14071497
APA StyleBeck, J. J., Ahmed, T., Finnicum, C. T., Zwinderman, K., Ehli, E. A., Boomsma, D. I., & Hottenga, J. J. (2023). Genetic Ancestry Estimates within Dutch Family Units and Across Genotyping Arrays: Insights from Empirical Analysis Using Two Estimation Methods. Genes, 14(7), 1497. https://doi.org/10.3390/genes14071497