
Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders
 , ,
, ,  ,
,  , , ,
, , ,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval and Sample Collection
2.2. Genetic Analysis
2.2.1. Whole-Exome Sequencing (WES)
2.2.2. Homozygosity Mapping
2.2.3. Sanger Sequencing
3. Results
3.1. Clinical Findings
- Family A
- Family B
- Family C
- Family D
- Family E
- Family F
- Family G
- Family H
- Family I
3.2. Molecular Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marsden, J.F. Cerebellar Ataxia. In Handbook of Clinical Neurology; Medimops: Berlin, Germany, 2018; Volume 159, pp. 261–281. [Google Scholar]
- Buckley, E.; Mazzà, C.; McNeill, A. A systematic review of the gait characteristics associated with Cerebellar Ataxia. Gait Posture 2018, 60, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Salinas, S.; Proukakis, C.; Crosby, A.; Warner, T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008, 7, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Suenaga, M.; Watanabe, H.; Sobue, G. Cognitive impairment in spinocerebellar degeneration. Eur. Neurol. 2009, 61, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Yahia, A.; Stevanin, G. The history of gene hunting in hereditary spinocerebellar degeneration: Lessons from the past and future perspectives. Front. Genet. 2021, 12, 638730. [Google Scholar] [CrossRef]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef]
- Parodi, L.; Coarelli, G.; Stevanin, G.; Brice, A.; Durr, A. Hereditary ataxias and paraparesias: Clinical and genetic update. Curr. Opin. Neurol. 2018, 31, 462–471. [Google Scholar] [CrossRef]
- Manto, M.; Gandini, J.; Feil, K.; Strupp, M. Cerebellar ataxias: An update. Curr. Opin. Neurol. 2020, 33, 150–160. [Google Scholar] [CrossRef]
- Synofzik, M.; Schüle, R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Yousaf, H.; Fatima, A.; Ali, Z.; Baig, S.M.; Toft, M.; Iqbal, Z. A Novel Nonsense Variant in GRM1 Causes Autosomal Recessive Spinocerebellar Ataxia 13 in a Consanguineous Pakistani Family. Genes 2022, 13, 1667. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, S.; Salpietro, V.; Malintan, N.; Poncelet, M.; Kriouile, Y.; Fortuna, S.; De Zorzi, R.; Payne, K.; Henderson, L.B.; Cortese, A.; et al. Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain 2019, 142, 2948–2964. [Google Scholar] [CrossRef] [PubMed]
- Klar, J.; Ali, Z.; Farooq, M.; Khan, K.; Wikström, J.; Iqbal, M.; Zulfiqar, S.; Faryal, S.; Baig, S.M.; Dahl, N. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur. J. Hum. Genet. 2017, 25, 848–853. [Google Scholar] [CrossRef] [PubMed]
- de Sainte Agathe, J.-M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Human Genom. 2023, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, M.; Peter, V.G.; Bedoni, N.; Bertrand, B.R.; Cisarova, K.; Salmaninejad, A.; Sepahi, N.; Rodrigues, R.; Piran, M.; Mojarrad, M.; et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 2021, 12, 518. [Google Scholar] [CrossRef]
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Tian, S.; Tariq, M.; Khan, S.; Safeer, M.; Ullah, N.; Akbar, N.; Javed, I.; Asif, M.; Ahmad, I.; et al. NGS-driven molecular diagnosis of heterogeneous hereditary neurological disorders reveals novel and known variants in disease-causing genes. Mol. Genet. Genom. 2022, 297, 1601–1613. [Google Scholar] [CrossRef]
- Nicolaou, P.; Georghiou, A.; Votsi, C.; Middleton, L.T.; Zamba-Papanicolaou, E.; Christodoulou, K. A novel c. 5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Med. Genet. 2008, 9, 28. [Google Scholar] [CrossRef]
- Algahtani, H.; Shirah, B.; Algahtani, R.; Naseer, M.I.; Al-Qahtani, M.H.; Abdulkareem, A.A. Ataxia with ocular apraxia type 2 not responding to 4-aminopyridine: A rare mutation in the SETX gene in a Saudi patient. Intractable Rare Dis. Res. 2018, 7, 275–279. [Google Scholar] [CrossRef]
- Gautam, R.; Sharma, M. Prevalence and diagnosis of neurological disorders using different deep learning techniques: A meta-analysis. J. Med. Syst. 2020, 44, 49. [Google Scholar] [CrossRef]
- Hamamy, H.; Antonarakis, S.E.; Cavalli-Sforza, L.L.; Temtamy, S.; Romeo, G.; Kate, L.P.T.; Bennett, R.L.; Shaw, A.; Megarbane, A.; van Duijn, C.; et al. Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet. Med. 2011, 3, 841–847. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Crimella, C.; Airoldi, G.; Polishchuk, R.; Bonato, S.; Brighina, E.; Scarlato, M.; Musumeci, O.; Toscano, A.; Martinuzzi, A.; et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013, 136, 3119–3139. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H. Lysosomal Dysfunctions in Hereditary Spastic Paraplegias, in Lysosomes-Associated Diseases and Methods to Study Their Function. IntechOpen 2017. [Google Scholar] [CrossRef]
- Bibi, F.; Efthymiou, S.; Bourinaris, T.; Tariq, A.; Zafar, F.; Rana, N.; Salpietro, V.; Houlden, H.; Raja, G.K.; Saeed, S.; et al. Rare novel CYP2U1 and ZFYVE26 variants identified in two Pakistani families with spastic paraplegia. J. Neurol. Sci. 2020, 411, 116669. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.; Monies, D.M.; Ramzan, K.; Hagos, S.; Bastaki, L.; Meyer, B.; Bohlega, S. Novel B4GALNT1 mutations in a complicated form of hereditary spastic paraplegia. Clin. Genet. 2014, 86, 500–501. [Google Scholar] [CrossRef]
- Boukhris, A.; Schüle-Freyer, R.; Loureiro, J.L.; Lourenço, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Lebrigio, R.F.A.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Harlalka, G.V.; Lehman, A.; Chioza, B.; Baple, E.L.; Maroofian, R.; Cross, H.; Sreekantan-Nair, A.; Priestman, D.A.; Al-Turki, S.; McEntagart, M.E.; et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013, 136, 3618–3624. [Google Scholar] [CrossRef]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.A.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef]
- Moreira, M.-C.; Klur, S.; Watanabe, M.; Németh, A.H.; Le Ber, I.; Moniz, J.-C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schöls, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef]
- Splinter, D.; Tanenbaum, M.E.; Lindqvist, A.; Jaarsma, D.; Flotho, A.; Yu, K.L.; Grigoriev, I.; Engelsma, D.; Haasdijk, E.D.; Keijzer, N.; et al. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010, 8, e1000350. [Google Scholar] [CrossRef]
- Neveling, K.; Martinez-Carrera, L.A.; Hölker, I.; Heister, A.; Verrips, A.; Hosseini-Barkooie, S.M.; Gilissen, C.; Vermeer, S.; Pennings, M.; Meijer, R.; et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am. J. Hum. Genet. 2013, 92, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef]
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.E. Molecular and cellular function of ALS2/alsin: Implication of membrane dynamics in neuronal development and degeneration. Neurochem. Int. 2007, 51, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.-C.; Barbot, C.; Tachi, N.; Kozuka, N.; Uchida, E.; Gibson, T.J.; Mendonça, P.; Costa, M.; Barros, J.; Yanagisawa, T.; et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001, 29, 189–193. [Google Scholar] [CrossRef]
- Date, H.; Onodera, O.; Tanaka, H.; Iwabuchi, K.; Uekawa, K.; Igarashi, S.; Koike, R.; Hiroi, T.; Yuasa, T.; Awaya, Y.; et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001, 29, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.A.; Kern, M.J.; Fullbright, G.; Bielawski, J.; Scherer, S.S.; Yum, S.W.; Li, J.J.; Cheng, H.; Han, X.; Venkata, J.K.; et al. Central nervous system dysfunction in a mouse model of FA2H deficiency. Glia 2011, 59, 1009–1021. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
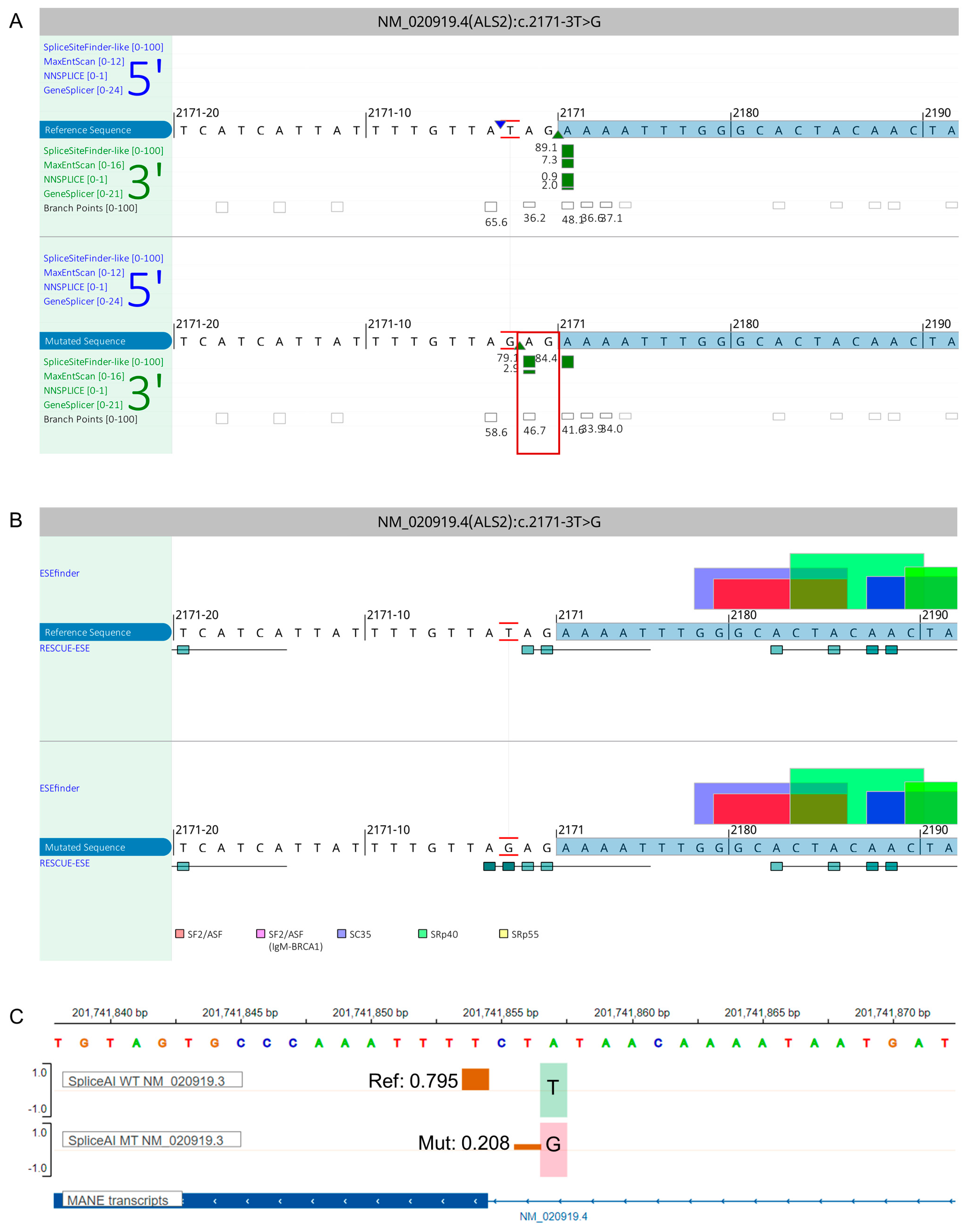{kind=link}
| Pedigree | Patient ID | Age (y) | Gender | Onset (y) | Clinical Diagnosis | Symptoms and Signs |
|---|---|---|---|---|---|---|
| Family A | IV:2 | 28 | M | N/A | Spastic paraplegia 15 | No ambulation, intellectual disability, muscle atrophy, dysarthria |
| IV:3 | 18 | F | N/A | Ataxic gait, intellectual disability, muscle atrophy, dysarthria, hip dysplasia | ||
| V:1 | 15 | F | N/A | Ataxic gait, intellectual disability, muscle atrophy | ||
| Family B | V:1 | N/A | F | N/A | Spastic Ataxia | Ataxic gait, delayed developmental milestones |
| V:2 | N/A | F | N/A | Ataxic gait, delayed developmental milestones | ||
| V:3 | N/A | M | N/A | Ataxic gait, delayed developmental milestones | ||
| Family C | V:1 | 17 | F | 6 | Spinal muscular atrophy, lower extremity-predominant, 2A, autosomal dominant | Gait abnormality, club feet, pes planus, short stature |
| V:2 | 11 | F | 4 | Gait abnormality, club feet, pes planus, hip dysplasia | ||
| V:3 | 12 | M | 4 | Gait abnormality, club feet, pes planus, hip dysplasia, upper limbs weakness, short stature, lower motor neuron deterioration symptoms | ||
| V:4 | 6 | M | 3 | No ambulation, club feet, lower motor neuron deterioration symptoms | ||
| Family D | V:1 | 24 | M | 1.5 | Infantile ascending hereditary spastic paraplegia | Muscle spasticity, Progressive loss of movement, bending of hands and feet, dysarthria, dysphagia, muscular atrophy, no cognitive deficit |
| V:5 | 14 | M | 5 | Muscle spasticity, Progressive loss of movement, bending of hands and feet, dysarthria, dysphagia, muscular atrophy, no cognitive deficit | ||
| Family E | V:4 | 22 | M | 2 | Infantile ascending hereditary spastic paraplegia | Muscle deterioration, progressive loss of movement, bending of hands and feet, dysarthria, dysphagia, hyper salivation, no cognitive deficit |
| V:5 | 25 | M | 2 | Muscle deterioration, progressive loss of movement, bending of hands and feet, dysarthria, dysphagia, hyper salivation, no cognitive deficit | ||
| VI:1 | 14 | M | 2 | Muscle deterioration, progressive loss of movement, bending of hands and feet, dysarthria, hyper salivation, no cognitive deficit | ||
| VI:2 | 6 | M | 2 | Muscle deterioration, progressive loss of movement, bending of hands and feet, hyper salivation, no cognitive deficit | ||
| VI:3 | 3 | M | 2 | Muscle deterioration, progressive loss of movement, hyper salivation, no cognitive deficit | ||
| Family F | IV:1 | 26 | M | 7 | Spastic paraplegia 26 | Ataxic gait, mild intellectual disability |
| IV:5 | 20 | M | 6 | Ataxic gait, dysarthria, mild intellectual disability | ||
| IV:6 | 15 | F | 6 | No ambulation, intellectual disability, dysarthria, in toeing | ||
| IV:7 | 10 | F | 8 | Ataxic gait, mild intellectual disability | ||
| Family G | V:1 | 10 | F | 3 | Spastic paraplegia 35 | No ambulation, hyperreflexia, lower limbs muscle atrophy |
| V:2 | 4 | F | 3 | Ataxic gait, hyperreflexia, lower limbs muscle atrophy | ||
| V:3 | 3 | M | 3 | Mild ataxic gait | ||
| Family H | V:1 | 40 | M | 16 | Ataxia with oculomotor apraxia type 1 | Progressive loss of ambulation, intellectual disability |
| V:2 | 37 | M | 15 | Progressive loss of ambulation, intellectual disability | ||
| V:3 | 34 | F | 16 | Progressive loss of ambulation, intellectual disability | ||
| V:4 | 51 | M | 16 | Progressive loss of ambulation, intellectual disability | ||
| Family I | V:1 | 32 | M | 12 | Ataxia with oculomotor apraxia type 2/ Spinocerebellar ataxia with axonal neuropathy 2 | Progressive ataxic gait, experienced febrile seizure till 8 years of age, action tremor, dysarthria, nystagmus |
| V:8 | 37 | M | 15 | Progressive ataxic gait, choreic movement, nystagmus | ||
| V:11 | 23 | F | 16 | Progressive ataxic gait, choreic movements, weak eyesight, fainting episodes |
| Family A | Family B | Family C | Family D | Family E | Family F | Family G | Family H | Family I | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Variant Annotation | Gene | ZFYVE26 | SACS | BICD2 | ALS2 | ALS2 | B4GALNT1 | FA2H | APTX | SETX |
| Transcript | ENST00000347230.9 | ENST00000682944.1 | ENST00000356884.10 | ENST00000264276.10 | ENST00000264276.11 | ENST00000341156.8 | ENST00000219368.8 | ENST00000379817.7 | ENST00000224140.6 | |
| GRCh38/hg38 position (DNA change) | 14:68272260 | 13:23355411 | 9:92717899 | 2:201741857 | 2:201726701 | 12:57631248 | 16:74774580 | 9:32984712 | 9:135187207 | |
| cDNA change | c.1093del | c.1201C>T | c.2156A>T | c.2171-3T>G | c.3145T>A | c.334_335dup | c.159_176del | c.689T>G | c.5308_5311del | |
| Protein change | p.Leu365SerfsTer16 | p.Arg401Ter | p.Lys719Met | N/A | p.Tyr1049Asn | p.Ala113GlyfsTer27 | p.53_58del | p.Val230Gly | p.Glu1770fs | |
| Variant type | Frameshift | Stop | Missense | Splice site | Missense | Frameshift | Inframe 18bp del | Missense | Frameshift | |
| Zygosity | Homozygous | Homozygous | Heterozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | |
| Allele Frequencies | dbSNP ID | N/A | rs769212398 | N/A | N/A | N/A | N/A | rs759947457 | rs536584919 | rs750959420 |
| gnomADv3 (highest subpopulation) | N/A | 0.00000658 | N/A | N/A | N/A | N/A | N/A | 0.0000132 | N/A | |
| gnomADv2.1 (highest subpopulation) | N/A | 0.00000398 | N/A | N/A | N/A | N/A | N/A | 0.0000159 | 0.0000318 | |
| Ensembl browser | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 0.0002 | N/A | |
| Iranome | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| GME Variome | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| ACMG Classification | Pathogenic (PVS1, PM2, PP1, PP4) | Pathogenic (PVS1, PM2, PP1, PP3,PP5) | Likely Pathogenic (PM1, PM2, PP1, PP2, PP3, PP4) | Uncertain Significance (PP1, PP3, PP4, PM2) | Uncertain Significance (PM2, PP1, PP3, PP4) | Pathogenic (PVS1, PM2, PP1, PP4) | Pathogenic (PS3, PM1, PM4, PM2, PP1, PP4) | Pathogenic (PS1, PM1, PM2, PP1, PP3, PP4) | Pathogenic (PVS1, PM2, PP1, PP4) | |
| Insilico Predictions | GERP++RS | N/A | 5.72 | N/A | N/A | 5.72 | N/A | N/A | 5.59 | N/A |
| CADD | N/A | 36 | 31 | 23.2 | 29 | N/A | N/A | 27.4 | 32 | |
| Polyphen-2 | N/A | N/A | 0.999 | N/A | 1 | N/A | N/A | 1 | N/A | |
| SIFT | N/A | N/A | 0 | N/A | 0 | N/A | N/A | 0 | N/A | |
| Mutation Taster | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saadi, S.M.; Cali, E.; Khalid, L.B.; Yousaf, H.; Zafar, G.; Khan, H.N.; Sher, M.; Vona, B.; Abdullah, U.; Malik, N.A.; et al. Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders. Genes 2023, 14, 1404. https://doi.org/10.3390/genes14071404
Saadi SM, Cali E, Khalid LB, Yousaf H, Zafar G, Khan HN, Sher M, Vona B, Abdullah U, Malik NA, et al. Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders. Genes. 2023; 14(7):1404. https://doi.org/10.3390/genes14071404
Chicago/Turabian StyleSaadi, Saadia Maryam, Elisa Cali, Lubaba Bintee Khalid, Hammad Yousaf, Ghazala Zafar, Haq Nawaz Khan, Muhammad Sher, Barbara Vona, Uzma Abdullah, Naveed Altaf Malik, and et al. 2023. "Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders" Genes 14, no. 7: 1404. https://doi.org/10.3390/genes14071404
APA StyleSaadi, S. M., Cali, E., Khalid, L. B., Yousaf, H., Zafar, G., Khan, H. N., Sher, M., Vona, B., Abdullah, U., Malik, N. A., Klar, J., Efthymiou, S., Dahl, N., Houlden, H., Toft, M., Baig, S. M., Fatima, A., & Iqbal, Z. (2023). Genetic Investigation of Consanguineous Pakistani Families Segregating Rare Spinocerebellar Disorders. Genes, 14(7), 1404. https://doi.org/10.3390/genes14071404