
A Study of the Genetic Structure of Hybrid Camels in Kazakhstan

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Quality Assessment
2.3. Whole Genome Sequencing
2.4. Sequencing Data Analysis
2.5. Genetic Structure Analysis
3. Results and Discussion
3.1. Whole Genome Sequencing and Data Filtering
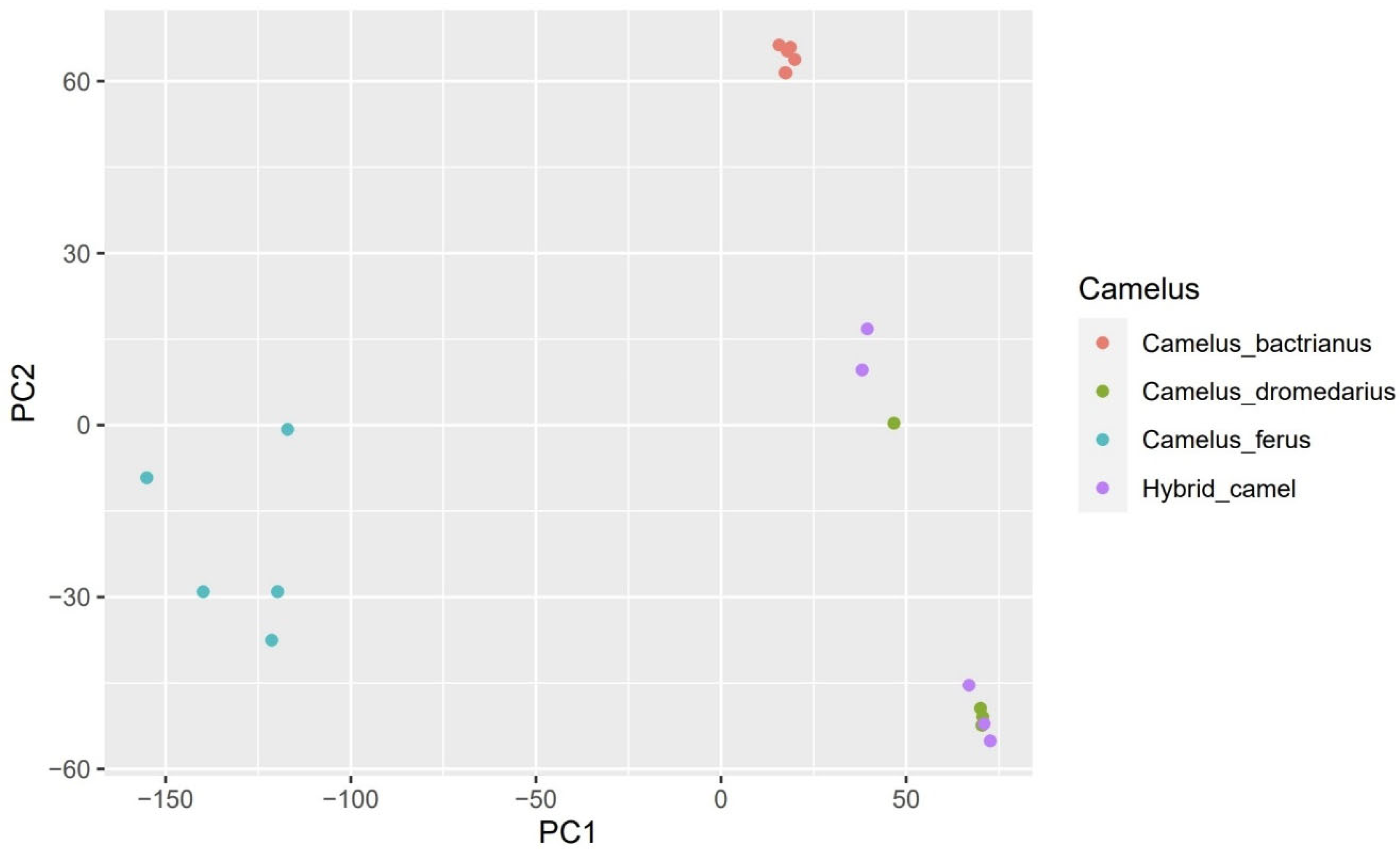
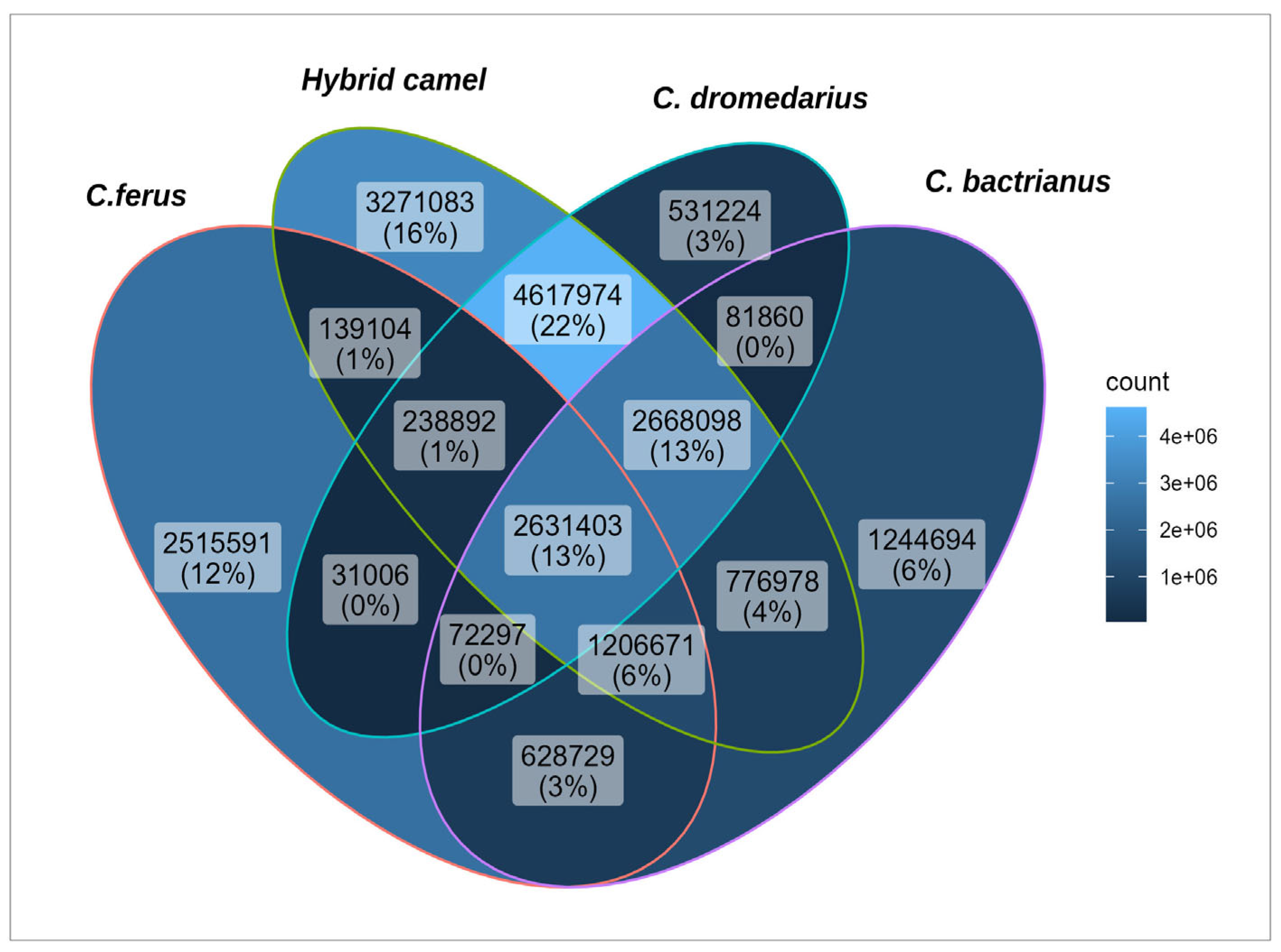
3.2. SNP Analyses
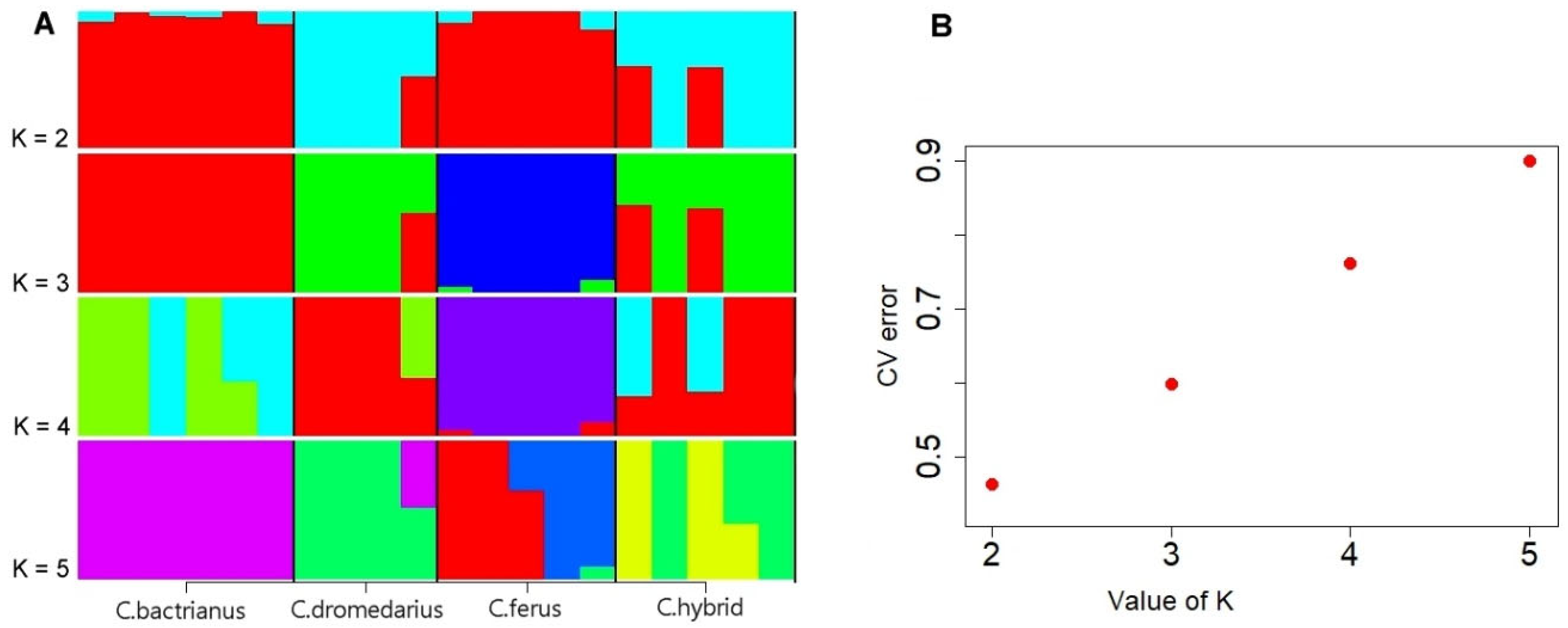
3.3. Population Structure Analysis
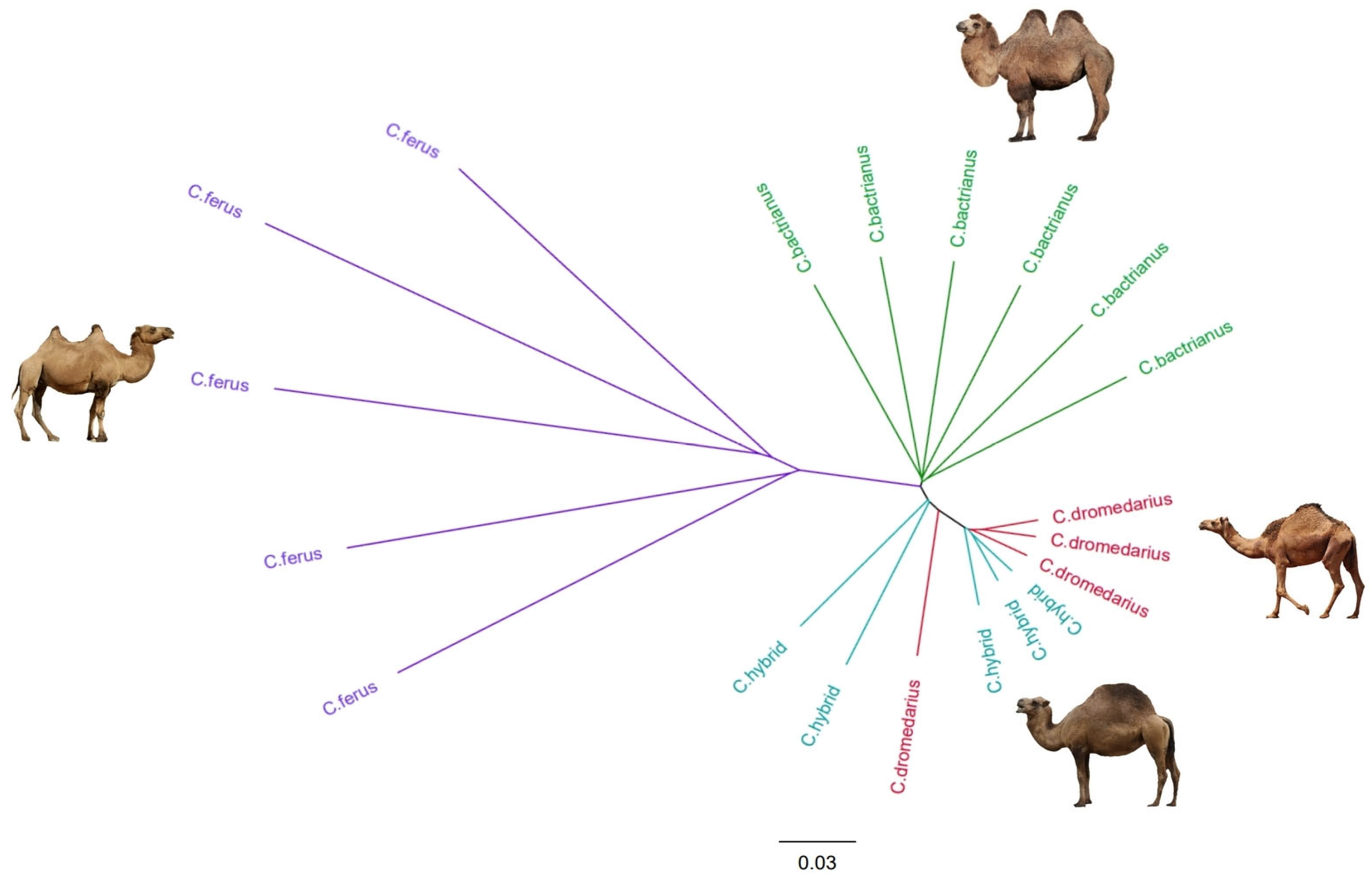
3.4. Phylogenetic Tree Construction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wapnish, P. Camel caravans and camel pastoralists at Tell Jemmeh. J. Anc. Near East. Soc. 1983, 13, 101–121. [Google Scholar]
- Balmus, G.; Trifonov, V.A.; Biltueva, L.S.; O’Brien, P.C.M.; Alkalaeva, E.S.; Fu, B.; Skidmore, J.A.; Allen, T.; Graphodatsky, A.S.; Yang, F. Cross-species chromosome painting among camel, cattle, pig and human: Further insights into the putative Cetartiodactyla ancestral karyotype. Chromosom. Res. 2007, 15, 499–514. [Google Scholar] [CrossRef]
- Kadwell, M.; Fernandez, M.; Stanley, H.F.; Baldi, R.; Wheeler, J.C.; Rosadio, R.; Bruford, M.W. Genetic analysis reveals the wild ancestors of the llama and the alpaca. Proc. R. Soc. Lond. 2001, 268, 2575–2584. [Google Scholar] [CrossRef]
- Ji, R.; Cui, P.; Ding, F.; Geng, J.; Gao, H.; Zhang, H.; Yu, J.; Hu, S.; Meng, H. Monophyletic origin of domestic bactrian camel (Camelus bactrianus) and its evolutionary relationship with the extant wild camel (Camelus bactrianus ferus). Anim. Genet. 2009, 40, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; AbuTarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef]
- Mohandesan, E.; Fitak, R.R.; Corander, J.; Yadamsuren, A.; Chuluunbat, B.; Abdelhadi, O.; Raziq, A.; Nagy, P.; Stalder, G.; Walzer, C.; et al. Mitogenome Sequencing in the Genus Camelus Reveals Evidence for Purifying Selection and Long-term Divergence between Wild and Domestic Bactrian Camels. Sci. Rep. 2017, 7, 9970. [Google Scholar] [CrossRef] [PubMed]
- Murray, E. Fowler Medicine and Surgery of South American Camelids: Llama Alpaca Vicuna Guanaco; Iowa State University Press: Ames, LA, USA, 1995. [Google Scholar]
- Dioli, M. Dromedary (Camelus dromedarius) and Bactrian camel (Camelus bactrianus) crossbreeding husbandry practices in Turkey and Kazakhstan: An in-depth review. Pastor. Res. Policy Pract. 2020, 10, 6. [Google Scholar] [CrossRef]
- Faye, B.; Konuspayeva, G.; Messad, S.; Loiseau, G. Discriminant milk components of Bactrian camel (Camelus bactrianus), dromedary (Camelus dromedarius) and hybrids. Dairy Sci. Technol. 2008, 88, 607–617. [Google Scholar] [CrossRef]
- Konuspayeva, G.; Faye, B.; Loiseau, G.; Levieux, D. Lactoferrin and immunoglobin content in camel milk from Kazakhstan. J. Dairy Sci. 2006, 90, 38–46. [Google Scholar] [CrossRef]
- Baimukanov, D.A. Cytogenetics and Selection of Two-Humped, One-Humped Camels and Their Hybrids; Bastau: Almaty, Kazakhstan, 2002; p. 160. [Google Scholar]
- Burger, P. The history of Old World camelids in the light of molecular genetics. Trop. Anim. Health Prod. 2016, 48, 905–913. [Google Scholar] [CrossRef]
- Wild Camels. Available online: https://www.wildcamels.com/bactrian-camels/ (accessed on 5 February 2023).
- Eldala. Available online: https://eldala.kz/novosti/zhivotnovodstvo/12959-pogolove-loshadey-v-kazahstane-vyroslo-na-12-za-god (accessed on 5 February 2023).
- Piro, M. Aspects of Molecular Genetics in Dromedary Camel. Front. Genet. 2021, 12, 723181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Levin, I.; Vallejo, R.L.; Khatib, H.; Dodgson, J.B.; Crittenden, L.B.; Hillel, J. Development of a genetic map of the chicken with markers of high utility. Poult. Sci. 1995, 74, 1855–1874. [Google Scholar] [CrossRef]
- Piro, M.; Mabsoute, F.E.; Khattaby, N.E.; Laghouaouta, H.; Boujenane, I. Genetic variability of dromedary camel populations based on microsatellite markers. Animal 2020, 14, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.H.; Abu-Tarbush, F.M.; Alshaik, M.; Aljumaah, R.; Saleh, A. Genetic diversity and population genetic structure of six dromedary camel (Camelus dromedarius) populations in Saudi Arabia. Saudi J. Biol. Sci. 2020, 27, 384–1389. [Google Scholar]
- Almathen, F.; Charruau, P.; Mohandesan, E.; Mwacharo, J.M.; Orozco-Terwengel, P.; Pitt, D.; Abdussamad, A.M.; Uerpmann, M.; Uerpmann, H.P.; De Cupere, B.; et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc. Natl. Acad. Sci. USA 2016, 113, 6707–6712. [Google Scholar] [CrossRef]
- Eynard, S.E.; Windig, J.J.; Hiemstra, S.J.; Calus, M.P.L. Whole-genome sequence data uncover loss of genetic diversity due to selection. Genet. Sel. Evol. 2016, 48, 33. [Google Scholar] [CrossRef]
- Lado, S.; Elbers, J.P.; Rogers, M.F.; Melo-Ferreira, J.; Yadamsuren, A.; Corander, J.; Horin, P.; Burger, P.A. Nucleotide Diversity of Functionally Different Groups of Immune Response Genes in Old World Camels Based on Newly Annotated and Reference-Guided Assemblies. BMC Genom. 2020, 21, 606. [Google Scholar] [CrossRef]
- Burger, P.A.; Palmieri, N. Estimating the Population Mutation Rate from a de novo Assembled Bactrian Camel Genome and Cross-Species Comparison with Dromedary ESTs. J. Hered. 2014, 105, 839–846. [Google Scholar] [CrossRef]
- Khalkhali-Evrigh, R.; Hafezian, S.H.; Hedayat-Evrigh, N.; Farhadi, A.; Bakhtiarizadeh, M.R. Genetic variants analysis of three dromedary camels using whole genome sequencing data. PLoS ONE 2018, 13, e0204028. [Google Scholar] [CrossRef]
- Ruiz, E.; Mohandesan, E.; Fitak, R.R.; Burger, P.A. Diagnostic Single Nucleotide Polymorphism Markers to Identify Hybridization Between Dromedary and Bactrian Camels. Conserv. Genet. Resour. 2015, 7, 329–332. [Google Scholar] [CrossRef]
- Bitaraf Sani, M.; Zare Harofte, J.; Banabazi, M.H.; Esmaeilkhanian, S.; Shafei Naderi, A.; Salim, N.; Teimoori, A.; Bitaraf, A.; Zadehrahmani, M.; Burger, P.A.; et al. Genomic Prediction for Growth Using a Low-Density SNP Panel in Dromedary Camels. Sci. Rep. 2021, 11, 7675. [Google Scholar] [CrossRef]
- Al-Sharif, M.M.; Radwan, H.A.; Hendam, B.M.; Ateya, A.I. Exploring single nucleotide polymorphisms in GH, IGF-I, MC4R and DGAT1 genes as predictors for growth performance in dromedary camel using multiple linear regression analysis. Small Rumin. Res. 2022, 207, 106619. [Google Scholar] [CrossRef]
- Ming, L.; Siren, D.; Hasi, S.; Jambl, T.; Ji, R. Review of genetic diversity in Bactrian camel (Camelus bactrianus). Anim. Front. 2022, 12, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.A.; Ciani, E. Structural and functional genomics in Old World camels—Where do we stand and where to go. Anim. Front. 2022, 12, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Akhmetsadykova, S.H.; Konuspayeva, G.; Akhmetsadykov, N. Camel breeding in Kazakhstan and future perspectives. Anim. Front. 2022, 12, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Adilbekova, E.; Alybayev, N.; Arunas, S.; Abuov, G. Genetic typing of South Kazakhstan populations’ dairy camels using DNA technology. Anim. Biotechnol. 2020, 31, 547–554. [Google Scholar]
- Amandykova, M.; Dossybayev, K.; Mussayeva, A.; Bekmanov, B.; Saitou, N. Comparative Analysis of the Polymorphism of the Casein Genes in Camels Bred in Kazakhstan. Diversity 2022, 14, 285. [Google Scholar] [CrossRef]
- Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 February 2023).
- Sourceforge.Net. Available online: https://bio-bwa.sourceforge.net/bwa.shtml (accessed on 5 February 2023).
- Htslib.Org. Available online: http://www.htslib.org/download/ (accessed on 5 February 2023).
- Github. Available online: https://broadinstitute.github.io/picard/ (accessed on 5 February 2023).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group, The variant call format and VCFtools. Bioinformatics 2011, 7, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- R-Project.Org. Available online: https://cran.r-project.org/web/packages/ggfortify/vignettes/plot_pca.html (accessed on 5 February 2023).
- Gao, C.-H.; Yu, G.; Cai, P. ggVennDiagram: An Intuitive, Easy-to-Use, and Highly Customizable R Package to Generate Venn Diagram. Front. Genet. 2021, 12, 706907. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Rambaut, A. FigTree v1.4.2 Molecular Evolution, Phylogenetics and Epidemiology, Institute of Evolutionary Biology, University of Edinburgh. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 February 2023).
- Hedayat-Evrigh, N.; Khalkhali-Evrigh, R.; Bakhtiarizadeh, M.R. Genome-Wide Identification and Analysis of Variants in Domestic and Wild Bactrian Camels Using Whole-Genome Sequencing Data. Int. J. Genom. 2020, 2020, 2430846. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Yuan, L.; Yi, L.; Ding, G.; Hasi, S.; Chen, G.; Jambl, T.; Hedayat-Evright, N.; Batmunkh, M.; Badmaevna, G.K.; et al. Whole-genome sequencing of 128 camels across Asia reveals origin and migration of domestic Bactrian camels. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef]
- Cog-Genomics.org. Available online: https://www.cog-genomics.org/plink/ (accessed on 5 February 2023).
- Sabahat, S.; Brauning, R.; Clarke, S.M.; Nadeem, A.; Thomson, P.C.; Khatkar, M.S. SNP discovery and population structure analysis in Lassi and Marecha camel breeds using a genotyping by sequencing method. Anim. Genet. 2020, 51, 620–623. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Description |
|---|---|
| Hybrid-1 | ♀ Bactrian × ♂ Dromedary |
| Hybrid-2 | ♀ Bactrian × ♂ Dromedary |
| Hybrid-3 | ♀ Dromedary × ♂ Bactrian |
| Hybrid-4 | ♀ Bactrian × ♂ Dromedary |
| Hybrid-5 | ♀ Bactrian × ♂ Dromedary |
| № | DNA Samples | DNA Concentration (ng/µL) | |
|---|---|---|---|
| NanoDrop One Data | Qubit Fluorometer Data | ||
| 1 | Hybrid-1 | 57.6 | 53.4 |
| 2 | Hybrid-2 | 67.8 | 60.4 |
| 3 | Hybrid-3 | 51.2 | 48.6 |
| 4 | Hybrid-4 | 69.1 | 62.1 |
| 5 | Hybrid-5 | 55.3 | 53.6 |
| Sample ID 1 | Total Read Bases (bp) 2 | Total Reads 3 | GC (%) 4 | AT (%) 5 | Q20 (%) 6 | Q30 (%) 7 |
|---|---|---|---|---|---|---|
| Hybrid camel-1 | 129,838,121,006 | 859,855,106 | 41.94 | 58.06 | 97.17 | 92.70 |
| Hybrid camel-2 | 137,538,079,710 | 910,848,210 | 41.92 | 58.08 | 97.32 | 93.02 |
| Hybrid camel-3 | 207,996,610,094 | 1,377,460,994 | 41.91 | 58.09 | 97.23 | 82.89 |
| Hybrid camel-4 | 108,267,988,144 | 717,006,544 | 42.01 | 57.99 | 97.38 | 93.22 |
| Hybrid camel-5 | 134,823,736,024 | 892,872,424 | 41.84 | 58.16 | 97.36 | 93.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amandykova, M.; Dossybayev, K.; Mussayeva, A.; Saitou, N.; Zhunusbayeva, Z.; Bekmanov, B. A Study of the Genetic Structure of Hybrid Camels in Kazakhstan. Genes 2023, 14, 1373. https://doi.org/10.3390/genes14071373
Amandykova M, Dossybayev K, Mussayeva A, Saitou N, Zhunusbayeva Z, Bekmanov B. A Study of the Genetic Structure of Hybrid Camels in Kazakhstan. Genes. 2023; 14(7):1373. https://doi.org/10.3390/genes14071373
Chicago/Turabian StyleAmandykova, Makpal, Kairat Dossybayev, Aizhan Mussayeva, Naruya Saitou, Zhazira Zhunusbayeva, and Bakytzhan Bekmanov. 2023. "A Study of the Genetic Structure of Hybrid Camels in Kazakhstan" Genes 14, no. 7: 1373. https://doi.org/10.3390/genes14071373
APA StyleAmandykova, M., Dossybayev, K., Mussayeva, A., Saitou, N., Zhunusbayeva, Z., & Bekmanov, B. (2023). A Study of the Genetic Structure of Hybrid Camels in Kazakhstan. Genes, 14(7), 1373. https://doi.org/10.3390/genes14071373