Abstract
Polycystic Kidney Diseases (PKDs) consist of a genetically and phenotypically heterogeneous group of inherited disorders characterized by numerous renal cysts. PKDs include autosomal dominant ADPKD, autosomal recessive ARPKD and atypical forms. Here, we analyzed 255 Italian patients using an NGS panel of 63 genes, plus Sanger sequencing of exon 1 of the PKD1 gene and MPLA (PKD1, PKD2 and PKHD1) analysis. Overall, 167 patients bore pathogenic/likely pathogenic variants in dominant genes, and 5 patients in recessive genes. Four patients were carriers of one pathogenic/likely pathogenic recessive variant. A total of 24 patients had a VUS variant in dominant genes, 8 patients in recessive genes and 15 patients were carriers of one VUS variant in recessive genes. Finally, in 32 patients we could not reveal any variant. Regarding the global diagnostic status, 69% of total patients bore pathogenic/likely pathogenic variants, 18.4% VUS variants and in 12.6% of patients we could not find any. PKD1 and PKD2 resulted to be the most mutated genes; additional genes were UMOD and GANAB. Among recessive genes, PKHD1 was the most mutated gene. An analysis of eGFR values showed that patients with truncating variants had a more severe phenotype. In conclusion, our study confirmed the high degree of genetic complexity at the basis of PKDs and highlighted the crucial role of molecular characterization in patients with suspicious clinical diagnosis. An accurate and early molecular diagnosis is essential to adopt the appropriate therapeutic protocol and represents a predictive factor for family members.
1. Introduction
Polycystic Kidney Diseases (PKDs) encompass a vast phenotypic spectrum of genetically and clinically heterogeneous disorders associated with different impacts on kidney function and with different treatment options [1,2].
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent among PKDs and the most common renal monogenic disease, affecting around 1 in 1000 individuals. ADPKD is a multisystemic disease with extrarenal manifestations, including polycystic liver disease (PLD), early onset hypertension, cerebral aneurysms and cardiovascular abnormalities and results the fourth cause of end-stage renal disease (ESRD) in adults [3,4,5]. Today, ADPKD is a condition treatable with tolvaptan, a selective vasopressin V2 receptor antagonist, and somatostatin analogues [6]. However, a differential diagnosis of ADPKD with other cystic renal diseases in certain cases remains crucial for early specific treatment.
Around 78% of ADPKD cases are linked to pathogenic variants in the PKD1 gene and around 15% to pathogenic variants in the PKD2 gene, while 7% of ADPKD cases remain genetically unresolved or are due to rare pathogenic variants in other genes [7,8]. Actually, about 2322 pathogenic variants in the PKD1 gene and 278 in the PKD2 gene distributed without preferential high spots are listed in the ADPKD Mutation Database at Mayo Clinic (https://pkdb.mayo.edu/variants).
PKD1 is a large complex human gene that spans 52 Kb on the 16p13.3 chromosome region and contains 46 exons; in addition, the 5′ to 33 exon region of PKD1 is replicated in six pseudogenes that share 98% homology [9]. PKD2, located on the 4q21 chromosome region, is a gene constituted by 15 exons [10]. The PKD1 and PKD2 genes encode Polycystin 1 (PC1) and 2 (PC2), respectively, two membrane glycoproteins with the primary cilium as a likely functional site [11]. Another important genetic cause of renal-related morbidity is the autosomal recessive form of PKDs (ARPKD) caused mainly by mutations in the PKHD1 gene, chromosome 6p12. PKHD1 is among the largest human genes, with 86 exons assembled into a variety of alternatively spliced transcripts with the longest continuous open reading frame that encodes a multi-domain integral membrane protein (fibrocystin/polyductin) of unknown function [12]. The PKHD1 mutations are also scattered throughout the gene and most of them are unique to single families [13].
The genetic heterogeneity of PKDs complicates the pathophysiology and the possibility of performing a precise and early diagnosis [14,15]. To further hamper the genetic diagnosis, it should be considered that (a) several other genetically inherited conditions have been associated with the formation of cysts in the kidney, and (b) there is a significant phenotypic and genotypic overlap between PKDs and polycystic liver diseases (PLD).
Genetic testing for PKD is a rapidly increasing area: applying PKD molecular testing in the clinical context could allow for diagnosis in suspected cases with no apparent family history or with equivocal imaging findings, improving patient care through disease prognostication, guide treatment, clinical trials and genetic counseling.
In this context, Next-Generation Sequencing (NGS) technology has upgraded the possibility to perform molecular diagnosis as well as to understand the genetic mechanisms linked to PKD conditions [16,17,18,19,20]. In this scenario, and based on literature data, we developed an NGS panel that includes 63 genes linked to PKDs to explore the genetic basis of the heterogeneous spectrum of PKD-like phenotypes in a large group of 255 Italian patients with suspicion or clinical diagnosis of PKD. We also performed Sanger sequencing of exon 1 of the PKD1 gene and MPLA analysis on patients whose pathogenic variants were not identified.
2. Materials and Methods
2.1. Patient Recruitment, Clinic and Biochemical Evaluation
In total, 255 consecutive patients were recruited from Nephrology Unit, Department of Public Health, “Federico II” University, Naples, and Hospital outpatient clinic, “Federico II”, Naples. All patients were 18 years of age or above, able to provide informed consent and without previous genetic results related to PKD. Patients included were either with a positive family history of PKD and met the unified criteria for ultrasonographic diagnosis of PKD [5] or were without PKD family history but had confirmed renal cysts through imaging as per the unified criteria for ultrasonographic diagnosis of PKD. A 10 mL blood sample was collected in EDTA tubes from each enrolled patient. Genomic DNA was isolated from each blood sample using the MagPurix Blood DNA Extraction kit (Zinexts Life Science Corp., New Taipei City, Taiwan). The study was approved by the local ethics committee and complied with the guidelines of the Declaration of Helsinki. Informed written consent was obtained from each patient.
2.2. NGS Custom Panel Design and Panel Content
Molecular testing was carried out by analyzing a panel of target genes through an NGS-based procedure. For the selection of genes (n = 63) included in the panel, we relied on updated data from the literature together with the advice received from nephrologists [16,17]. The complete design of the PKDs panel is available in Supplementary Table S1. This panel includes several classes of genes involved in (i) folding, trafficking or degradation of kidney proteins; (ii) the protection of kidney epithelial cells; (iii) renal tubulogenesis; (iv) primary cilia formation and maturation; and (v) related syndromes. For each gene, we analyzed the coding regions, 25 bp in each of the intronic boundaries.
2.3. DNA Isolation and NGS Library Preparation and Sequencing
DNA reference samples (n = 26) were obtained from the hospital “Casa Sollievo della Sofferenza, Istituto di Ricovero e Cura a Carattere Scientifico”, San Giovanni Rotondo, Foggia, Italy, and from CEINGE-Biotecnologie Avanzate “Franco Salvatore”, Napoli, Italy. Genomic DNA (gDNA) was isolated using the Nucleon BACC3 Genomic DNA Extraction Kit (GE Healthcare, Life Sciences, Chicago, IL, USA) according to the manufacturer’s instructions. The quality of DNA samples was assessed by the TapeStation system (Agilent Technologies, Santa Clara, CA, USA); a DNA integrity number (DIN) >6 was considered suitable for NGS analysis. DNA quantity was evaluated through the NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and by using Qubit dsDNA BR and HS assays kits (Life Technologies, Carlsbad, CA, USA).
DNA molecular analysis was performed using the above-mentioned NGS panel. HaloPlex technology (Agilent, Santa Clara, CA, USA) was used for library preparation. In detail, each genomic DNA sample was fragmented using a pool of restriction enzymes. The obtained fragments were enriched with hybridization with the custom capture probes, and then purified and PCR-amplified to obtain a DNA library or sample (total probes: 42,817; total probes size: 415.055 kbp; each probe was 120 mer). During this procedure, each genomic DNA sample was univocally tagged with a barcode sequence to allow for sample multiplexing during the subsequent sequencing step. The custom design of our probes was realized using the web-based SureDesign application. A total of 50 ng of gDNA was processed through the SureSelectQXT Target Enrichment system (Agilent Technologies, Santa Clara, CA, USA) for Illumina multiplexed sequencing. Briefly, gDNA was enzymatically fragmented and adaptor-tagged to obtain a pool of fragments that were amplified by PCR reaction. Then, the prepared DNA library amplicons were hybridized to the capture custom library, made up of 63 selected genes, and purified by streptavidin-coated magnetic beads. The captured, target-enriched DNA library was amplified by PCR reaction by using dual index primers, which allowed us to univocally barcode each sample. Finally, SureSelect-enriched dual-indexed NGS samples were pooled together for multiplexed sequencing. The sequencing reactions were carried out on the MiSeq instrument (Illumina, San Diego, CA, USA) using a flow cell micro, running 6/8 samples for each sequencing run to obtain an average coverage of about 100× (>90% of the analyzable target regions were covered by at least 50×).
2.4. NGS Data Analysis
The Alissa Align & Call v1.0.2.10 tool (Agilent Technologies, Santa Clara, CA, USA), using the genome build hg38 as a reference, was used to perform alignments, variant calling and quality filtering. The median QV bases used in variant calling was 39, with an average read length of 141 bp. Variant filtering and interpretation were performed using Alissa Interpret v5.2.6 CE IVD software (Agilent Technologies, Santa Clara, CA, USA), using GRCh38.p2 and annotation sources such as 1000 Genomes (Phase 3 release v5, 10 September 2014, including GRCh38 data), ClinVar (NCBI ClinVar October 2019), DGV (Database of Genomic Variants, version 15 May 2016), ESP6500 (variants in the ESP6500SI-V2 dataset of the exome sequencing project, annotated with SeattleSeqAnnotation137), ExAC (ExAC release 1.0—including GRCh38 from lift over data) and OMIM (OMIM, version 25 October 2019). To define mutations as novel, we also checked for their presence in two large public sequence databases: gnomAD (release 2.0.2, available at gnomad.broadinstitute.org; n = 15.496 diploid genome sequences and n = 123.136 diploid exome sequences) and BRAVO (powered by TOPMed Freeze5 on genome reference consortium human build 38 patch release 12, available at https://bravo.sph.umich.edu/freeze5/hg38/; n = 62.784 diploid whole-genome sequences).
2.5. Sanger Sequencing
All the variants considered, such as pathogenic, likely pathogenic and VUS, were confirmed by Sanger sequencing from genomic DNA extracted from the second patient’s blood sample. PCR amplification was carried out on a 2720 Thermal Cycler (Applied Biosystems Inc., Foster City, CA, USA) or VeritiPro Thermal Cycler (Applied Biosystems Inc., Foster City, CA, USA). Next, direct sequencing was performed using a 3730 DNA Analyzer (Applied Biosystem, Foster City, CA, USA). In particular, for the exons 1–46 of PKD1, we performed a nested PCR of the region of interest from LR amplicons before proceeding to Sanger sequencing. For the VUS and/or LP variants in PKD1 gene with a low read depth (<20%), we performed targeted genotyping using specific primers (designed on the canonic gene or on the pseudogenes) to confirm that the functional PKD1 gene was amplified and sequenced rather than the pseudogene. Sanger sequencing of exon 1 of the PKD1 gene was performed as previously described only on those patients negative for pathogenic variants (primers are available on request) [18].
2.6. Multiplex Ligation-Dependent Probe Amplification (MLPA)
After NGS procedure, genomic DNA from patient samples without variants classified as pathogenic, likely pathogenic or VUS was analyzed by MLPA for the detection of large gene rearrangements using the commercial kits SALSA MLPA P351 PKD1 and SALSA MLPA P352 PKD1-PKD2 probemix (MRC-Holland, Amsterdam, The Netherlands). Successively, for those patients without positive results in PKD1-PKD2 SALSA MLPA, we proceeded to analyzing the PKHD1 gene for the detection of large gene rearrangements using the commercial SALSA MLPA P341-B4 PKHD1 mix 1 and P342-C1 kits PKHD1 mix 2 probemix kits (MRC-Holland, Amsterdam, NL).
PCR product analysis was carried out on a 3730 DNA Analyzer and the electropherograms were visualized by Coffalyzer software version 130202.2357 (MRC Holland, Amsterdam, The Netherlands). The assays were performed according to the manufacturer’s instructions.
2.7. Variant’s Pathogenicity Predictions
Bioinformatics predictions of the variant’s effects were performed using the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Clinvar tools (https://www.ncbi.nlm.nih.gov/clinvar/). Further predictions were assessed with the Mutation Taster tool (http://www.mutationtaster.org) and VarSome website (https://varsome.com/variant/hg38). All software were used with their default parameters. Variant classification was performed according to American College of Medical Genetics and Genomics (ACMG) guidelines [21]. Variants were classified into five-tier categories: pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign and benign.
The standard gene variant nomenclature, according to Human Genome Variation Society (http://www.hgvs.org/mutnomen) and corrected by https://mutalyzer.nl, was used. Information on protein structure and domains was obtained by consulting Uniprot (http://www.uniprot.org) and the PFam web resources (http://pfam.xfam.org/).
3. Results
In this study, we analyzed a large cohort of 249 Caucasian and 6 non-Caucasian patients affected by PKDs through a targeted NGS gene panel consisting of 63 genes related to PKD genetic diseases; 225 patients were unrelated and 30 were related. We found 167 patients bearing pathogenic/likely pathogenic variants in dominant genes and 5 patients with recessive genes of heterozygous or homozygous status; in 4 patients, we found only one pathogenic/likely pathogenic recessive variant. A total of 24 patients had a VUS variant in dominant genes while 8 patients had a variant in recessive genes; in 15 patients we found only one VUS variant in recessive genes. Finally, in 32 patients, both NGS and MLPA did not reveal any pathogenic mutation. The complete list of patients and genes involved is reported in Table 1.

Table 1.
Genetic classification of PKD population.
Regarding the global diagnostic status distribution, 69% of the total identified variants in our cohort of patients were pathogenic/likely pathogenic variants (Supplementary Tables S2 and S4), 18.4% were VUS variants (Supplementary Tables S3 and S5), and in 12.6% of patients we could not find any variants, see Figure 1, panel A. The majority of those pathogenic/likely pathogenic variants were found in dominant genes and, when we analyzed the involved genes, we found that, as expected, PKD1 and PKD2 were the most mutated ones, see Figure 1, panel B.
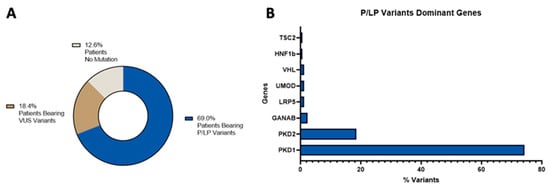
Figure 1.
Distribution of the molecular diagnostic status (panel A) and of the pathogenic/likely pathogenic variants in dominant genes (panel B).
Nonsense and missense mutations were the most frequent pathogenic/likely pathogenic variants type within PKD1, accounting for 37.19% and 20.66%, respectively, of total PKD1-mutated patients; interestingly, among PKD2-mutated patients, the nonsense mutations represented the most frequent mutations (60% of total pathogenic/likely pathogenic PKD2 variants). Phenotypically, we observed that PKD2-mutated patients showed an eGFR of 56.75 vs. 73.76 PKD1 patients (p = 0.028). However, the age at diagnosis of PKD1 patients (birth–64 years old, median 25) was lower than PKD2 (20–60 years old, median 42) suggesting that PKD2 patients are diagnosed at an older age compared to PKD1.
We identified, in the PKD1 gene, 83 different pathogenic/likely pathogenic variants and 16 VUS variants were found (see Supplementary Tables S2 and S4), 56% of the former were located in the exons 15, 23 and 45 (Figure 2, panel A). It can be noticed that exon 15, together with 5, 13 and 14, within PKD1 encodes for the PKD globular domain of the Polycystin-1 (PC1) protein that represents a crucial structural and functional element of PC1 being involved in interactions with other proteins (cell–cell or cell–matrix interactions). On the other hand, the exons 23 and 44 were also frequently found to be mutated. These two latter exons are involved in the production of the PC1 C-terminal cytoplasmic tail, a fundamental domain for the interaction between PC1 and PC2 proteins, both crucial in renal disease progression.
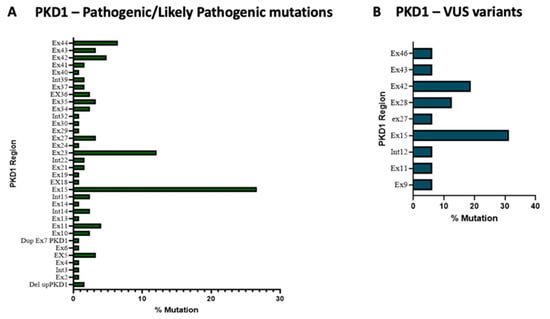
Figure 2.
Exon’s pathogenic/likely pathogenic variants (panel A) and VUS variants frequency in PKD1 gene (panel B).
Regarding the VUS variants within PKD1, exons 15 and 42 are the most frequently mutated, accounting for 44.3% of total variants (Figure 2, panel B).
Nineteen different pathogenic/likely variants and two VUS variants were found within the PKD2 gene (Supplementary Tables S2 and S4). Exons 5 and 14 of PKD2 accounted for the majority of pathogenic/likely pathogenic variants (41.93%) (see Figure 3, panel A); interestingly, exon 14 encodes the COOH-terminal region through which the transmembrane glycoprotein polycystin-2 (PC2) interacts with PC-1. Regarding the VUS variants within PKD2, we found only two variants in exon 2 and intron 14 (see Figure 3, panel B).
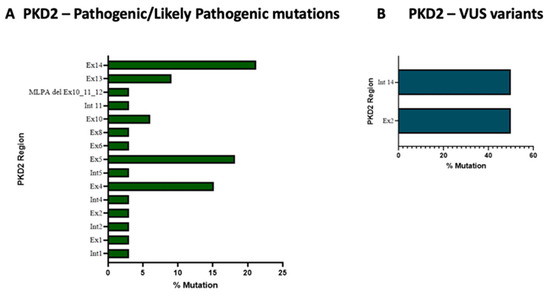
Figure 3.
Exons’ pathogenic/likely pathogenic variants (panel A) and VUS variants’ frequency in PKD2 gene (panel B).
Among PKD1-positive patients, we found a family bearing a likely pathogenic variant in PKD1 (c.10549G>T) together with a concomitant variant in the α-galactosidase A gene (GLA) (c.868A>C). Deficiency of the lysosomal GLA enzyme causes Fabry disease (OMIM: 301500), a rare X-linked hereditary disease (incidence 1:117.000 live births). The first patient to come to our attention was a 31-year old female patient with renal cysts and an eGFR of 92: NGS analysis revealed the presence of both the above-mentioned mutations. Successively, two relatives (one brother and a sister) were analyzed: the brother had both variants in GLA and PKD1 with a moderate renal impairment plus a cardiac and skin impairment from Fabry disease, while the sister had the PKD1 variant only. NGS analysis of the mother (with normal renal function) revealed that she is carrier of the GLA mutation. The father is PKD1-mutated with a severe PKD clinical phenotype.
For those patients without variants classified as pathogenic, likely pathogenic or VUS, we proceeded by analyzing exon 1 by Sanger sequencing and successively analyzing large gene rearrangements by MLPA (Figure 4). No mutations were found in exon 1. MPLA analysis identified three patients with large rearrangements in PKD1: two with large deletions (Figure 4, panels A,C) and one with duplication (Figure 4, panel B). In addition, one patient had a large deletion in PKD2 (Figure 4, panel D).
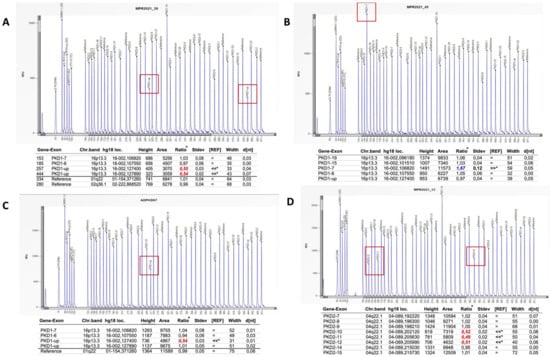
Figure 4.
MPLA analysis has revealed two PKD1 deletions (panels A,C), one PKD1 duplication (panel B) and one deletion in PKD2 gene (panel D).
The genotype description of the five recessive patients is shown in Table 2. Four of them reported two variants in PKHD1 while one patient was homozygous for MAPKBP1. Two PKHD1 patients and the MAPKBP1-positive patient bore pathogenic/likely pathogenic variants, while the other PKHD1 patients carried VUS variants. It can be noticed that, within the entire cohort of patients, the MAPKBP1-positive patient was the only homozygous genotype we found. The mean eGFR of ARPKD patients was 62 vs. 72.3 for ADPKD ones.
Table 2.
Genotype description of ARPKD recessive patients (compound heterozygous).
Trans-heterozygous patients with two variants in different recessive genes are reported in Table 3. Only two patients carried pathogenic mutations, while in the other patients we could only identify VUS variants.
Table 3.
Genotype description of PKD patients with two variants in different recessive genes (trans-heterozygous patients).
NGS analysis revealed that 101 patients had truncating mutations in the PKD1, PKD2, PKHD1 and MAPKBP1 genes. The mean e-GFR of patients bearing truncating mutations was 62.97 (73.58 for PKD1 patients, 52.35 for PKD2 patients, 67 for PKDH1 patients and 9 for MAPKBP1 patients, respectively) vs. 70.42 for all remaining patients (Table 4). Of note, all truncating mutations are predicted to be pathogenic or likely pathogenic, confirming that these mutations strongly affect protein structure and function.
Table 4.
Genotype description of truncating mutations.
We found 37 not previously reported pathogenic/likely pathogenic alterations: 31 in PKD1; 4 in PKD2; 1 in UMOD; and 1 in MAPKB1 genes (see Table 5). Of them, 16.2% were missense mutations, 45.9% were frameshift and 10.8% were splicing. In addition to those variants, we also found 15 VUS novel variants whose clinical meaning needs to be confirmed by further analyses.
Table 5.
Genotype description of patients bearing novel pathogenic/likely pathogenic variants.
4. Discussion
In this study, we analyzed a large cohort of 255 Italian PKD patients through a targeted NGS gene panel composed of 63 genes related to PKD genetic diseases. We found 167 patients carrying pathogenic/likely pathogenic variants in the dominant genes. Five patients had pathogenic/probably pathogenic variants in the recessive genes: four in the heterozygous state and one was homozygous in the MAPKBP1 gene. Finally, in four patients, we found only one pathogenic/likely pathogenic variant. As regards the VUS variants, 24 patients presented a VUS variant in the dominant genes while 8 patients in the recessive genes; in 15 patients we found only one VUS variant in the recessive genes. Finally, in 32 patients, both NGS and MLPA analyses did not reveal any variants.
Genetics is presently rarely used for routine diagnostics in PKDs because there are many issues hampering the genetic diagnosis of these diseases: the large size, the high genetic complexity and the lack of mutational hotspot in the causative genes. However, in recent years, NGS technology has proven to be an effective alternative to conventional and unconventional techniques in PKDs [36,37,38]. Here, we described the design and development of an NGS panel (PKDs panel) to simultaneously screen the coding regions (CDS) of the genes together with 25 bp of regions adjacent to both the 5′ and 3′ of the 63 genes. With the NGS technique plus Sanger sequencing and MLPA, we describe here a methodological approach able to perform the molecular diagnosis of about 70% of patients with clinical evidence of PKD.
Considering the Italian population, previous studies analyzed ADPKD patients, reaching an overall detection rate of maximum 80% [7,16,39,40]. Another Italian group considered 119 individuals with inherited kidney diseases (polycystic and non-polycystic), testing 115 genes causing renal diseases and identifying the disease-causing variants in 51.5% and 40% of polycystic and non-polycystic individuals, respectively [40]. All of these data suggest that early clinical differentiation into cystic and non-cystic kidney disease is essential for best outcomes [41].
However, in cases of unclear or negative family anamnesis, on the other hand, it may be necessary to modify the clinical diagnosis, essentially based on imaging, following the genetic diagnosis; in fact, the same clinical phenotype may be due to mutations present in other PKD-associated genes. Therefore, knowing the genotype becomes very important in these cases to make the right therapeutic decisions.
In fact, for several patients (both affected by recessive forms as well as affected by dominant forms, but carriers of mutations in genes other than PKD1–2), conservative or target therapy (tolvaptan/octreotide) is not appropriate and/or even ineffective. Therefore, a wrong clinical diagnosis could potentially create a serious delay in the correct therapeutic approach, preventing personalized patient management.
Here, we identified in the PKD1 gene 83 different pathogenic/likely pathogenic variants and 16 VUS variants, 56% of which were located in exons 15, 23 and 45. It can be seen that exon 15, together with 5, 13 and 14, within PKD1 encodes for the PKD globular domain of the Polycystin-1 (PC1) protein that represents a crucial structural and functional element of PC1 being involved in interactions with other proteins (cell–cell or cell–matrix interactions). On the other hand, exons 23 and 42 were also frequently mutated in our patient population. These two latter exons are involved in the production of the PC1 C-terminal cytoplasmic tail, a fundamental domain for the interaction between PC1 and PC2 proteins, both crucial in renal disease progression [42].
In all patients found to be mutation-negative for the genes included in the NGS panel, we performed Sanger sequencing of PKD1 exon 1 (to obtain better and more complete sequencing) as well as MLPA analysis. This latter evaluation led to the identification of one large duplication and two large deletions in three patients with no mutations identified at the NGS procedure. Such data are in accordance with the frequency of large rearrangements reported by Carrera et al. [39]. From our analysis, it is interesting to outline that we found two patients—belonging to the same family—bearing a likely pathogenic mutation in PKD1 and a concomitant likely pathogenic mutation in the GLA gene. In detail, the family consists of the mother, asymptomatic with normal renal function, carrying the mutation in the GLA gene; the father, characterized by a severe renal phenotype, carrying the PKD1 mutation; and the three children—one daughter, with a normal kidney function, carrying the PKD1 mutation, a second daughter carrying both PKD1 and GLA mutations with normal renal function, mild proteinuria and absence of symptoms of Fabry disease, and a third brother carrying both PKD1 and GLA mutations, with moderate renal insufficiency along with cardiac and skin impairment from Fabry disease.
Based on the current clinical features, it is not possible to define whether there is a worsening of renal function in the male patient carrying both mutations, since they are not comparable to those of females. To our knowledge, only one published paper reported a similar genetic status [43]. The authors described a 60-year-old male patient who was diagnosed with Fabry disease at 34 years of age and a VUS variant in PKD1. In accordance with our observation, the patient fulfilled the clinical criteria for PKD. This observation further supports the importance of the genetic analysis of renal cystic patients through the examination of a large panel of PKD-associated genes. The presence of this patient in the PKD population confirms a precise indication of the importance of including the GLA gene in an NGS panel for the genetic diagnosis of PKD. Indeed, the formation of renal cysts in Fabry disease patients may represent an undiagnosed case of PKD [44].
To date, 278 mutations have been found in PKD2 (https://pkdb.mayo.edu/variants) and here we observed 19 different pathogenic/likely variants and 2 VUS variants were found within the PKD2 gene (Supplementary Tables S2 and S4). The exons 5 and 14 of PKD2 accounted for the majority of pathogenic/likely pathogenic variants (41.93%). Interestingly, the exon 14 encodes the COOH-terminal region through which the transmembrane glycoprotein polycystin-2 (PC2) interacts with PC-1. In particular, PC-2 forms homotetrameric transient receptor potential channels that regulate the intracellular calcium concentration from the endoplasmic reticulum membrane and the primary cilium membrane [45]. The molecular changes in the interaction between PKD1 and PKD2 genes increase cell proliferation and fluid secretion, mechanisms at the basis of cyst formation [45]. It is therefore quite relevant that exon 14 is the main site of the causative mutations of PKD2. The MLPA analysis of PKD2 led to the identification of one large deletion.
Interestingly, we found two unrelated ADPKD patients with pathogenic/likely pathogenic variants in LRP5 and two patients with LRP5 VUS variants. LRP5 is a co-receptor strongly expressed in both the liver and kidneys and involved in the Wnt signaling pathway, whose disruption may predominantly lead to polycystic liver disease [46,47,48]. LRP5 was first reported in relation to autosomal dominant polycystic liver disease with or without kidney cysts in 2014 [46]. Some expression studies have demonstrated LRP5 expression in liver tissue [46,47,48]. In addition, some studies in the literature have reported that, in up to 94% of patients, LRP5 variants may render ADPKD patients more susceptible to the development of polycystic liver disease [46,47,48]. Furthermore, there are functional and animal studies that support that LRP5 variants may contribute to hepatic and renal cystogenesis.
However, overall, there is limited and unconvincing evidence to support a gene–disease relationship between LRP5 and autosomal dominant polycystic liver disease with or without kidney cysts. Although more evidence is needed to support a causal role for polycystic disease, evidence has emerged that contradicts the gene–disease relationship.
In this scenario, our data are quite interesting considering that the clinical picture of our patients correlates with cystic kidney disease, suggesting that the LRP5 gene, in addition to polycystic liver disease, could potentially play a role in PKD predisposition.
Another gene causative of ADPKD is GANAB [49]. In this study, we identified the missense c.2087 G>A mutation (p.Arg696Gln) previously described (https://varsome.com/variant/hg38/ganab%20c.2087G%3EA?annotation-mode=germline) that co-segregates with the disease in four affected family members. The patients in this family suffered from both polycystic kidney as well as liver disease. In our cohort, 101 patients out of 257 had truncating mutations in PKD1, PKD2, PKHD1 and MAPKBP1. The mean e-GFR of those patients was 62.97 vs. 70.42 for all other patients with other types of mutation, confirming that truncating mutations are associated with a more severe phenotype. In accordance with our data, both Heyer et al., and Hwang et al., reported that truncating PKD1 mutations (frameshift, splicing and nonsense) have a more severe disease prognosis with lower eGFR [50,51,52,53].
Regarding the recessive form of the disease, PKHD1 resulted to be the most mutated gene, representing 34.61% of total alleles: four patients bore the canonical heterozygous genotype, while in four patients we were able to identify a single mutation in heterozygosity, suggesting that some variants in PKHD1 might cause ADPKD as well as ADPLD, as suggested by literature data [54,55]. However, the identification of a single mutation in PKHD1 might also be due to the lack of identification of the second causative variant. In addition, family members should be recruited to deepen understanding of the pathological role of such mutations.
Thirty-seven not previously reported pathogenic or likely pathogenic variants in four different genes were identified. Moreover, the elevated number of not previously reported variants confirms the elevated molecular heterogeneity of the disease within our Italian PKD population, whereas previous reports described novel mutations only in a few genes [16]. Finally, we identified only one VUS variant in heterozygosity in 15 patients. It can be noted that the pathogenicity of VUS variants, as well as the dominant or recessive transmission of some genes and genetic variants, rapidly evolve through additional studies; therefore, some VUS will likely be reclassified as disease-causing and more monogenic causes of kidney disease will be identified. In light of this possibility, the rate of positive findings of the study may likely be an underestimate. The missing mutations in heterozygous subjects and in patients without identified mutations could be located in deep intronic or other regulatory regions distant from the splice donor and acceptor sites that have not been screened so far; alternatively, causative mutations could reside in other genes not analyzed in the present study and, more importantly, still not identified as genes involved in cystic renal disease. It should be considered, indeed, that the molecular pathways underlying cystic formation in the kidneys are quite complex and involve hundreds of proteins. In addition, patients without evidenced mutations should be considered not only as molecular undiagnosed patients but also as patients that, although phenotypically adherent to the PKD picture, need to be considered as different subjects requiring a different clinical management [38,56,57]. For those patients, further studies are required to deepen the germline and somatic genetic background—also possibly including additional genes in the analysis [58].
The limitations of this study include a lack of some clinical information and a clinical follow-up, mainly due the non-compliance of most families. Another limitation of the study may be the design of the gene panel that, although inclusive of numerous genes related to polycystic disease, may not contain additional genes potentially responsible for PKD. Despite these limitations, our results confirm that the NGS-based panel approach is a useful first-line tool for PKDs diagnosis. In the near future, as more patients are investigated, the information provided by such panels will continue to grow and improve understanding of these complex pathologies and hopefully improve the efficiency of such investigations.
Furthermore, thanks to genetic analysis, it has been possible to carry out diagnoses in complex families (i.e., PKD1-GLA-mutated patients); cases in which the molecular results have had a decisive impact on management and correct therapeutic decisions.
Therefore, genetic analysis can help to reach a timely diagnosis, which is extremely important as it can put an end to diagnostic uncertainty and allow for appropriate disease-specific counseling and the implementation of personalized medical plans in accordance with the current disease-specific consensus guidelines and, in the longer term, it can support the development of better designed clinical trials [59].
Future clinical studies including larger cohorts of patients, the identification of additional genes, follow-up information and comparative analysis of healthy controls are needed. It is worth noting that in the PKD1 gene, the presence of six pseudogenes is a complicating issue in NGS, because their sequence can interfere with the detection of canonical variants and generate false-positive and false-negative variants; here, we have tried to overcome this issue using specific primers designed to exclude DNA regions belonging to the six pseudogenes [46]. Additional technologies such as Whole-Exome Sequencing (WES) or Whole-Genome Sequencing (WGS) could provide an alternative type of analysis or a second-step analysis of those patients in whom the mutation cannot be identified through NGS analysis. However, it should be noted that there are some limitations which make these approaches not easily feasible, such as the much longer turnaround times of WES and WGS to complete the diagnostic process together with the very high cost that do not always make the choice of these technological platforms appropriate. Molecular analysis will help to confirm the diagnosis in clinically uncertain/atypical cases, exclude the presence of a variant in donors for kidney transplantation for living donors, and offer genetic counselling for at-risk families [16,23,31,46,47].
5. Conclusions
In conclusion, our study confirms the high degree of genetic heterogeneity within the Italian population, further endorsing that several pathogenic mutations in different genes can be the molecular basis of the same clinical phenotype of PKD patients. An accurate and prompt but more importantly early molecular diagnosis is essential to adopt timely lifestyle choices and might also inform reproductive counselling, and could also be interesting as a predictive factor for family members. Currently, the greater chances of positive PKD clinical trial outcomes and lower drug dropouts are closely linked to a more precise and specific molecular diagnosis, highlighting the key role of molecular characterization in PKDs. Finally, molecular analysis helped to exclude the presence of a variant in donors for kidney transplantation and to offer genetic counselling for at-risk families.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/genes14061236/s1, Table S1: Genes comprised in the PKD disease NGS panel; Table S2: Pathogenic and likely pathogenic mutations in AD genes; Table S3: Pathogenic and likely pathogenic mutations in AR genes; Table S4: VUS variants in AD genes; Table S5: VUS alterations in AR genes.
Author Contributions
E.N. and A.D. conceived the study. D.D. and E.N. performed the genetic analysis. M.A., G.S., O.D.M., M.G., E.R. and A.P. recruited the patients and collected clinical information. A.D. and E.N. wrote the manuscript. A.D. and A.P. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Università della Campania “Vanvitelli”, SCAM Rice.Base.GiovaniRicercatori2022.AGAPAO Study.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee university of Naples “Federico II” (protocol code 2018-000477-77, date of approval: 16 November 2022.
Informed Consent Statement
Written informed consent was obtained from the patient(s).
Data Availability Statement
Not applicable.
Acknowledgments
We thank Alessandro Vito Lasorsa for making his bioinformatic expertise available for the analysis of the gnomAD allele frequency. We also thank “Fondazione Casa della Speranza” for having made resources available for this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fujimaru, T.; Mori, T.; Sekine, A.; Mandai, S.; Chiga, M.; Kikuchi, H.; Ando, F.; Mori, Y.; Nomura, N.; Iimori, S.; et al. Kidney enlargement and multiple liver cyst formation implicate mutations in PKD1/2 in adult sporadic polycystic kidney disease. Clin. Genet. 2018, 94, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Iliuta, I.-A.; Kalatharan, V.; Wang, K.; Gall, E.C.-L.; Conklin, J.; Pourafkari, M.; Ting, R.; Chen, C.; Borgo, A.C.; He, N.; et al. Polycystic Kidney Disease without an Apparent Family History. J. Am. Soc. Nephrol. 2017, 28, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Suwabe, T.; Shukoor, S.; Chamberlain, A.M.; Killian, J.M.; King, B.F.; Edwards, M.; Senum, S.R.; Madsen, C.D.; Chebib, F.T.; Hogan, M.C.; et al. Epidemiology of Autosomal Dominant Polycystic Kidney Disease in Olmsted County. Clin. J. Am. Soc. Nephrol. 2020, 15, 69–79. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Capuano, I.; Buonanno, P.; Riccio, E.; Rizzo, M.; Pisani, A. Tolvaptan vs. somatostatin in the treatment of ADPKD: A review of the literature. Clin. Nephrol. 2022, 97, 131–140. [Google Scholar] [CrossRef]
- Orisio, S.; Noris, M.; Rigoldi, M.; Bresin, E.; Perico, N.; Trillini, M.; Donadelli, R.; Perna, A.; Benigni, A.; Remuzzi, G. Mutation Analysis of PKD1 and PKD2 Genes in a Large Italian Cohort Reveals Novel Pathogenic Variants Including A Novel Complex Rearrangement. Nephron, 2023; online ahead of print. [Google Scholar] [CrossRef]
- Schönauer, R.; Baatz, S.; Nemitz-Kliemchen, M.; Frank, V.; Petzold, F.; Sewerin, S.; Popp, B.; Münch, J.; Neuber, S.; Bergmann, C.; et al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet. Med. 2020, 22, 1374–1383. [Google Scholar] [CrossRef]
- Bogdanova, N.; Markoff, A.; Gerke, V.; McCluskey, M.; Horst, J.; Dworniczak, B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics 2001, 74, 333–341. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Audrézet, M.P.; Renaudineau, E.; Hourmant, M.; Charasse, C.; Michez, E.; Frouget, T.; Vigneau, C.; Dantal, J.; Siohan, P.; et al. PKD2-Related Autosomal Dominant Polycystic Kidney Disease: Prevalence, Clinical Presentation, Mutation Spectrum, and Prognosis. Am. J. Kidney Dis. 2017, 70, 476–485. [Google Scholar] [CrossRef]
- Luo, L.; Roy, S.; Li, L.; Ma, M. Polycystic kidney disease: Novel insights into polycystin function. Trends Mol. Med. 2023, 29, 268–281. [Google Scholar] [CrossRef]
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2018, 5, 221. [Google Scholar] [CrossRef]
- Losekoot, M.; Haarloo, C.; Ruivenkamp, C.; White, S.J.; Breuning, M.H.; Peters, D.J. Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum. Genet. 2005, 118, 185–206. [Google Scholar] [CrossRef]
- Sekine, A.; Hidaka, S.; Moriyama, T.; Shikida, Y.; Shimazu, K.; Ishikawa, E.; Uchiyama, K.; Kataoka, H.; Kawano, H.; Kurashige, M.; et al. Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). J. Clin. Med. 2022, 11, 6528. [Google Scholar] [CrossRef] [PubMed]
- Snoek, R.; van Jaarsveld, R.H.; Nguyen, T.Q.; Peters, E.D.J.; Elferink, M.G.; Ernst, R.F.; Rookmaaker, M.B.; Lilien, M.R.; Spierings, E.; Goldschmeding, R.; et al. Genetics-first approach improves diagnostics of ESKD patients younger than 50 years. Nephrol. Dial. Transplant. 2022, 37, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, V.; Bin, S.; Graziano, C.; Capelli, I.; Minardi, R.; Aiello, V.; Ambrosini, E.; Cristalli, C.P.; Mattiaccio, A.; Pariali, M.; et al. Gene Panel Analysis in a Large Cohort of Patients with Autosomal Dominant Polycystic Kidney Disease Allows the Identification of 80 Potentially Causative Novel Variants and the Characterization of a Complex Genetic Architecture in a Subset of Families. Front. Genet. 2020, 11, 464. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, A.J.; Westemeyer, M.; Xie, J.; Bloom, M.S.; Brossart, K.; Eckel, J.J.; Jones, F.; Molnar, M.Z.; Kotzker, W.; Anand, P.; et al. Genetic Etiologies for Chronic Kidney Disease Revealed through Next-Generation Renal Gene Panel. Am. J. Nephrol. 2022, 53, 297–306. [Google Scholar] [CrossRef]
- Mochizuki, T.; Teraoka, A.; Akagawa, H.; Makabe, S.; Akihisa, T.; Sato, M.; Kataoka, H.; Mitobe, M.; Furukawa, T.; Tsuchiya, K.; et al. Mutation analyses by next-generation sequencing and multiplex ligation-dependent probe amplification in Japanese autosomal dominant polycystic kidney disease patients. Clin. Exp. Nephrol. 2019, 23, 1022–1030. [Google Scholar] [CrossRef]
- Renkema, K.Y.; Stokman, M.F.; Giles, R.H.; Knoers, N.V. Next-generation sequencing for research and diagnostics in kidney disease. Nat. Rev. Nephrol. 2014, 10, 433–444. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.-H.; Chang, C.L.; Song, S.H.; Kim, N. Novel PKD1 Mutations in Patients with Autosomal Dominant Polycystic Kidney Disease. Lab. Med. 2021, 52, 174–180. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesth. Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Thong, M.K.; Fietz, M.; Nicholls, C.; Lee, M.H.; Asma, O. Congenital disorder of glycosylation type Ia in a Malaysian family: Clinical outcome and description of a novel PMM2 mutation. J. Inherit. Metab. Dis. 2009, 32, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Stekrova, J.; Reiterova, J.; Svobodova, S.; Kebrdlova, V.; Lnenicka, P.; Merta, M.; Viklicky, O.; Kohoutova, M. New mutations in the PKD1 gene in Czech population with autosomal dominant polycystic kidney disease. BMC Med. Genet. 2009, 10, 78. [Google Scholar] [CrossRef]
- Dong, K.; Liu, X.; Jia, X.; Miao, H.; Ji, W.; Wu, J.; Huang, Y.; Xu, L.; Zhang, X.; Su, H.; et al. Disease causing property analyzation of variants in 12 Chinese families with polycystic kidney disease. Mol. Genet. Genom. Med. 2020, 8, e1467. [Google Scholar] [CrossRef]
- Tan, Y.C.; Blumenfeld, J.D.; Anghel, R.; Donahue, S.; Belenkaya, R.; Balina, M.; Parker, T.; Levine, D.; Leonard, D.G.; Rennert, H. Novel method for genomic analysis of PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease. Hum. Mutat. 2009, 30, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Jilg, C.; Bacher, J.; Nabulsi, Z.; Malinoc, A.; Hummel, B.; Hoffmann, M.M.; Ortiz-Bruechle, N.; Glasker, S.; Pisarski, P.; et al. Epidemiology of autosomal-dominant polycystic kidney disease: An in-depth clinical study for south-western Germany. Nephrol. Dial. Transplant. 2013, 28, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Blanco, F.D.V.; Trepiccione, F.; Blasio, G.; Torella, A.; Melluso, A.; Capolongo, G.; Pollastro, R.M.; Piluso, G.; Di Iorio, V.; et al. Nephroplex: A kidney-focused NGS panel highlights the challenges of PKD1 sequencing and identifies a founder BBS4 mutation. J. Nephrol. 2021, 34, 1855–1874. [Google Scholar] [CrossRef]
- Xu, D.; Ma, Y.; Gu, X.; Bian, R.; Lu, Y.; Xing, X.; Mei, C. Novel Mutations in the PKD1 and PKD2 Genes of Chinese Patients with Autosomal Dominant Polycystic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 297–309. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef]
- Domingo-Gallego, A.; Pybus, M.; Bullich, G.; Furlano, M.; Ejarque-Vila, L.; Lorente-Grandoso, L.; Ruiz, P.; Fraga, G.; González, M.L.; Piñero-Fernández, J.A.; et al. Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol. Dial. Transplant. 2022, 37, 687–696. [Google Scholar] [CrossRef]
- Robinson, C.; Hiemstra, T.F.; Spencer, D.; Waller, S.; Daboo, L.; Karet Frankl, F.E.; Sandford, R.N. Clinical utility of PKD2 mutation testing in a polycystic kidney disease cohort attending a specialist nephrology out-patient clinic. BMC Nephrol. 2012, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Viribay, M.; Hayashi, T.; Tellería, D.; Mochizuki, T.; Reynolds, D.M.; Alonso, R.; Lens, X.M.; Moreno, F.; Harris, P.C.; Somlo, S.; et al. Novel stop and frameshifting mutations in the autosomal dominant polycystic kidney disease 2 (PKD2) gene. Hum. Genet. 1997, 101, 229–234. [Google Scholar] [CrossRef]
- Trujillano, D.; Bullich, G.; Ossowski, S.; Ballarín, J.; Torra, R.; Estivill, X.; Ars, E. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol. Genet. Genom. Med. 2014, 2, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couvé, S.; et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Shuster, S.; Keunen, J.; Shannon, P.; Watkins, N.; Chong, K.; Chitayat, D. Prenatal detection of isolated bilateral hyperechogenic kidneys: Etiologies and outcomes. Prenat. Diagn. 2019, 39, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Strmecki, L.; Gamble, V.; Burton, S.; Sneddon, V.; Peral, B.; Roy, S.; Bakkaloglu, A.; Komel, R.; Winearls, C.G.; et al. Mutation analysis of the entire PKD1 gene: Genetic and diagnostic implications. Am. J. Hum. Genet. 2001, 68, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann. Intern. Med. 2018, 168, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Mallawaarachchi, A.C.; Ben Lundie, B.; Hort, Y.; Schonrock, N.; Senum, S.R.; Gayevskiy, V.; Minoche, A.E.; Hollway, G.; Ohnesorg, T.; Hinchcliffe, M.; et al. Genomic diagnostics in polycystic kidney disease: An assessment of real-world use of whole-genome sequencing. Eur. J. Hum. Genet. 2021, 29, 760–770. [Google Scholar] [CrossRef]
- Carrera, P.; Calzavara, S.; Magistroni, R.; Dunnen, J.T.D.; Rigo, F.; Stenirri, S.; Testa, F.; Messa, P.; Cerutti, R.; Scolari, F.; et al. Deciphering Variability of PKD1 and PKD2 in an Italian Cohort of 643 Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD). Sci. Rep. 2016, 6, 30850. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef]
- Johar, L.; Lee, G.; Martin-Rios, A.; Hall, K.; Cheng, C.; Lombardo, D.; Pahl, M.; Kimonis, V. Polycystic kidney disease complicates renal pathology in a family with Fabry disease. Mol. Genet. Metab. Rep. 2022, 33, 100934. [Google Scholar] [CrossRef]
- Pisani, A.; Daniele, A.; Di Domenico, C.; Nigro, E.; Salvatore, F.; Riccio, E. Late diagnosis of Fabry disease caused by a de novo mutation in a patient with end stage renal disease. BMC Res. Notes 2015, 8, 711. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef]
- Cnossen, W.R.; Morsche, R.H.M.T.; Hoischen, A.; Gilissen, C.; Chrispijn, M.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Veltman, J.A.; Drenth, J.P.H. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5343–5348. [Google Scholar] [CrossRef]
- Cnossen, W.R.; Drenth, J.P.H. Polycystic liver disease: An overview of pathogenesis, clinical manifestations and management. Orphanet J. Rare Dis. 2014, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Cnossen, W.R.; te Morsche, R.H.; Hoischen, A.; Gilissen, C.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Losekoot, M.; Breuning, M.H.; Peters, D.J.; et al. LRP5 variants may contribute to ADPKD. Eur. J. Hum. Genet. 2016, 24, 237–242. [Google Scholar] [CrossRef]
- Van De Laarschot, L.F.M.; Morsche, R.H.M.T.; Hoischen, A.; Venselaar, H.; Roelofs, H.M.; Cnossen, W.R.; Banales, J.M.; Roepman, R.; Drenth, J.P.H. Novel GANAB variants associated with polycystic liver disease. Orphanet J. Rare Dis. 2020, 15, 302. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.-H.; Conklin, J.; Chan, W.; Roslin, N.M.; Liu, J.; He, N.; Wang, K.; Sundsbak, J.L.; Heyer, C.M.; Haider, M.; et al. Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1861–1868. [Google Scholar] [CrossRef]
- Gall, E.C.-L.; Audrézet, M.-P.; Chen, J.-M.; Hourmant, M.; Morin, M.-P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P.; et al. Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Hussain, N.; Naim, M.; Zayed, M.; Al-Mulla, F.; Kehinde, E.O.; Seaburg, L.M.; Sundsbak, J.L.; Harris, P.C. A novel PKD1 variant demonstrates a disease-modifying role in trans with a truncating PKD1 mutation in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2015, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Haghighi, A.; di Bari, I.; Song, X.; Pei, Y. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin. J. Am. Soc. Nephrol. 2021, 16, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; LaRusso, N.F. Therapeutic Targets in Polycystic Liver Disease. Curr. Drug Targets 2017, 18, 950–957. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.-A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef]
- Capuano, I.; Buonanno, P.; Riccio, E.; Amicone, M.; Pisani, A. Therapeutic advances in ADPKD: The future awaits. J. Nephrol. 2022, 35, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Pezone, I.; Amicone, M.; Capuano, I.; Buonanno, P.; Riccio, E.; Pisani, A. Familial polycystic kidneys with no genetic confirmation: Are we sure it is ADPKD? Clin. Nephrol. 2022, 99, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, K.; Dm, Z.S.; Kerr, P.G.; Gaff, C.; Martyn, M.; Whitlam, J.; Creighton, B.; Bn, E.D.; Hunter, M.; Jarmolowicz, A.; et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet. Med. 2021, 23, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.X.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease Therapies on the Horizon. Adv. Kidney Dis. Health 2023, 30, 245–260. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, J.; Qiu, W.; Liang, C.; Li, C.; Wei, T.; Feng, Z.; Guo, Q.; Yang, K.; Liu, Z. Comprehensive strategy improves the genetic diagnosis of different polycystic kidney diseases. J. Cell. Mol. Med. 2021, 25, 6318–6332. [Google Scholar] [CrossRef]
- Thomas, C.P.; Daloul, R.; Lentine, K.L.; Gohh, R.; Anand, P.M.; Rasouly, H.M.; Sharfuddin, A.A.; Schlondorff, J.; Rodig, N.; Freese, M.E.; et al. Genetic evaluation of living kidney donor candidates: A review and recommendations for best practices. Am. J. Transplant. 2023, 23, 597–607. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).