
Digenic Congenital Hypogonadotropic Hypogonadism Due to Heterozygous GNRH1 p.R31C and AMHR2 p.G445_L453del Variants


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Case Report
3. Methods
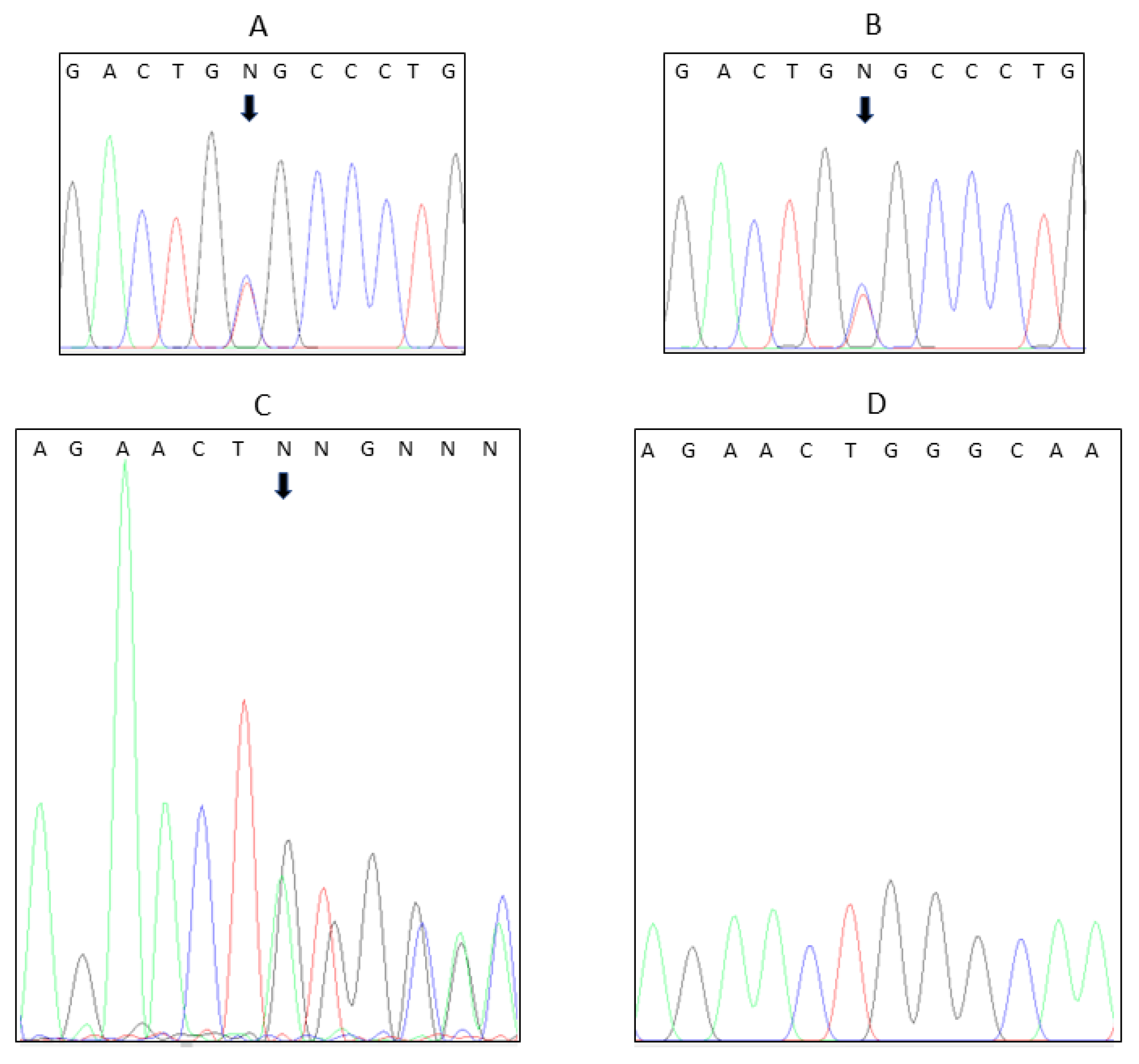
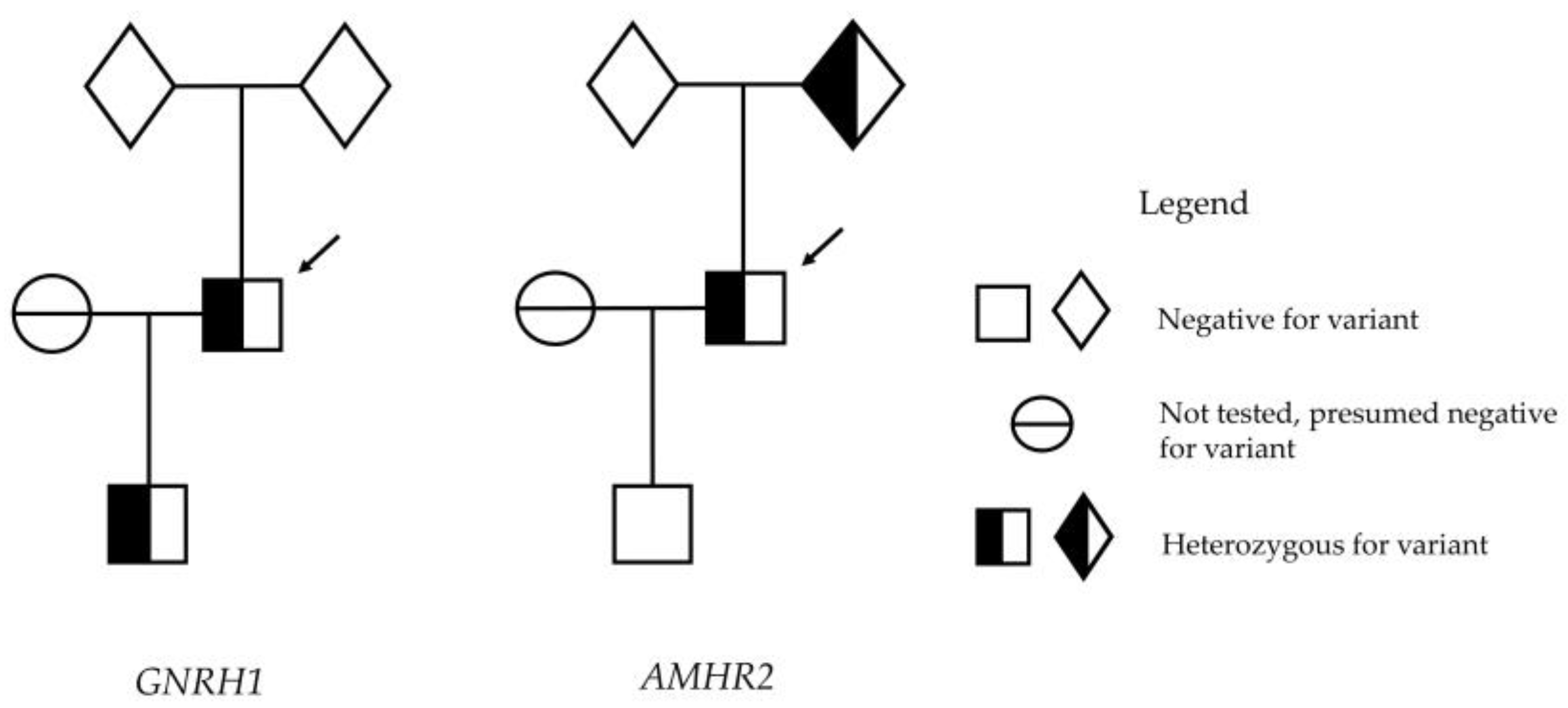
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cangiano, B.; Swee, D.S.; Quinton, R.; Bonomi, M. Genetics of congenital hypogonadotropic hypogonadism: Peculiarities and phenotype of an oligogenic disease. Hum. Genet. 2021, 140, 77–111. [Google Scholar] [CrossRef]
- Palmert, M.R.; Chan, Y.M.; Dunkel, L. Puberty and its disorders in the male. In Sperling Pediatric Endocrinology, 5th ed.; Elsevier: Philadelphia, PA USA, 2021; pp. 661–694. [Google Scholar]
- Kalfa, N.; Gaspari, L.; Ollivier, M.; Philibert, P.; Bergougnoux, A.; Paris, F.; Sultan, C. Molecular genetics of hypospadias and cryptorchidism recent developments. Clin. Genet. 2019, 95, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Vizeneux, A.; Hilfiger, A.; Bouligand, J.; Pouillot, M.; Brailly-Tabard, S.; Bashamboo, A.; McElreavey, K.; Brauner, R. Congenital hypogonadotropic hypogonadism during childhood: Presentation and genetic analyses in 46 boys. PLoS ONE 2013, 8, e77827. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, C.; Qin, M.; Liu, Y.; Tian, Y. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism. Clin. Endocrinol. 2017, 87, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamaki, J.; Raivio, T.; Pitteloud, N. Clinical management of congenital hypogonadotropic hypogonadism. Endocr. Rev. 2019, 40, 669–710. (In English) [Google Scholar] [CrossRef]
- Makela, J.A.; Koskenniemi, J.J.; Virtanen, H.E.; Toppari, J. Testis development. Endocr. Rev. 2019, 40, 857–905. [Google Scholar] [CrossRef]
- Dwyer, A.A.; Jayasena, C.N.; Quinton, R. Congenital hypogonadotropic hypogonadism: Implications of absent mini-puberty. Minerva Endocrinol. 2016, 41, 188–195. [Google Scholar] [PubMed]
- Chan, Y.M.; de Guillebon, A.; Lang-Muritano, M.; Plummer, L.; Cerrato, F.; Tsiaras, S.; Gaspert, A.; Lavoie, H.B.; Wu, C.H.; Crowley, W.F., Jr.; et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2009, 106, 11703–11708. [Google Scholar] [CrossRef] [PubMed]
- Quaynor, S.D.; Kim, H.G.; Cappello, E.M.; Williams, T.; Chorich, L.P.; Bick, D.P.; Sherins, R.J.; Layman, L.C. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 2011, 96, 1424–1430.e6. [Google Scholar] [CrossRef]
- Maione, L.; Albarel, F.; Bouchard, P.; Gallant, M.; Flanagan, C.A.; Bobe, R.; Cohen-Tannoudji, J.; Pivonello, R.; Colao, A.; Brue, T.; et al. R31C GNRH1 mutation and congenital hypogonadotropic hypogonadism. PLoS ONE [Electron. Resour.] 2013, 8, e69616. [Google Scholar]
- PGD (Implementation) Technical Advisory Committee. Policy on Approval of Diagnostic Procedures Involving Embryos; Reproductive Technology Council, Department of Health, Western Australia: Perth, Australia, 2017. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, J.; Maloney, K.A.; Pollin, T.I.; Jeng, L.J. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 1165. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef]
- Sunyaev, S.; Ramensky, V.; Koch, I.; Lathe, W., 3rd; Kondrashov, A.S.; Bork, P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001, 10, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Swee, D.S.; Quinton, R. Congenital hypogonadotrophic hypogonadism: Minipuberty and the case for neonatal diagnosis. Front. Endocrinol. 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Bouligand, J.; Ghervan, C.; Tello, J.A.; Brailly-Tabard, S.; Salenave, S.; Chanson, P.; Lombes, M.; Millar, R.P.; Guiochon-Mantel, A.; Young, J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Engl. J. Med. 2009, 360, 2742–2748. [Google Scholar] [CrossRef]
- Mengen, E.; Tunc, S.; Kotan, L.D.; Nalbantoglu, O.; Demir, K.; Gurbuz, F.; Turan, I.; Seker, G.; Yuksel, B.; Topaloglu, A.K. Complete Idiopathic Hypogonadotropic Hypogonadism due to Homozygous GNRH1 Mutations in the Mutational Hot Spots in the Region Encoding the Decapeptide. Horm. Res. Paediatr. 2016, 85, 107–111. [Google Scholar] [CrossRef]
- Amato, L.G.L.; Montenegro, L.R.; Lerario, A.M.; Jorge, A.A.L.; Guerra Junior, G.; Schnoll, C.; Renck, A.C.; Trarbach, E.B.; Costa, E.M.F.; Mendonca, B.B.; et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2019, 181, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Sagi, S.V.; Joshi, H.; Whiles, E.; Hikmat, M.; Puthi, V.R.; MacDougall, J.; Spiden, S.L.; Fuller, G.; Park, S.M.; Oyibo, S.O. Normosmic idiopathic hypogonadotropic hypogonadism due to a novel GNRH1 variant in two siblings. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 19–0145. [Google Scholar]
- Brachet, C.; Gernay, C.; Boros, E.; Soblet, J.; Vilain, C.; Heinrichs, C. Homozygous p.R31H GNRH1 mutation and normosmic congenital hypogonadotropic hypogonadism in a patient and self-limited delayed puberty in his relatives. J. Pediatr. Endocrinol. Metab. 2020, 33, 1237–1240. [Google Scholar] [CrossRef]
- Turkyilmaz, A.; Cayir, A.; Yarali, O.; Kurnaz, E.; Kartal Baykan, E.; Arslan Ates, E.; Demirbilek, H. Clinical characteristics and molecular genetic analysis of a cohort with idiopathic congenital hypogonadism. J. Pediatr. Endocrinol. Metab. 2021, 34, 771–780. [Google Scholar] [PubMed]
- Patil, V.A.; Lila, A.R.; Shah, N.L.N.; Ekbote, A.V.; Shah, R.; Bhandare, V.V.; Sarathi, V.; Arya, S.; Memon, S.S.; Kunwar, A.; et al. GNRH1 variants in congenital hypogonadotropic hypogonadism: Single-center experience and systematic literature review. Neuroendocrinology 2022, 112, 723–732. [Google Scholar] [CrossRef]
- Cassatella, D.; Howard, S.R.; Acierno, J.S.; Xu, C.; Papadakis, G.E.; Santoni, F.A.; Dwyer, A.A.; Santini, S.; Sykiotis, G.P.; Chambion, C.; et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur. J. Endocrinol. 2018, 178, 377–388. [Google Scholar] [CrossRef]
- Zhou, C.; Niu, Y.; Xu, H.; Li, Z.; Wang, T.; Yang, W.; Wang, S.; Wang, D.W.; Liu, J. Mutation profiles and clinical characteristics of Chinese males with isolated hypogonadotropic hypogonadism. Fertil. Steril. 2018, 110, 486–495.e5. [Google Scholar] [CrossRef]
- Acierno, J.S.; Xu, C.; Papadakis, G.E.; Niederlander, N.J.; Rademaker, J.D.; Meylan, J.; Messina, A.; Kolesinska, Z.; Quinton, R.; Lang-Muritano, M.; et al. Pathogenic mosaic variants in congenital hypogonadotropic hypogonadism. Genet. Med. 2020, 22, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Leung, P.C. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev. 2005, 26, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P.; Flanagan, C.A.; Milton, R.C.; King, J.A. Chimeric analogues of vertebrate gonadotropin-releasing hormones comprising substitutions of the variant amino acids in positions 5, 7, and 8. Characterization of requirements for receptor binding and gonadotropin release in mammalian and avian pituitary gonadotropes. J. Biol. Chem. 1989, 264, 21007–21013. [Google Scholar] [PubMed]
- Lu, Z.; Coetsee, M.; White, C.; Millar, R. Structural determinants for ligand-receptor conformational selection in a peptide G protein-coupled receptor. J. Biol. Chem. 2007, 282, 17921–17929. [Google Scholar] [CrossRef]
- Malone, S.A.; Papadakis, G.E.; Messina, A.; Mimouni, N.E.H.; Trova, S.; Imbernon, M.; Allet, C.; Cimino, I.; Acierno, J.; Cassatella, D.; et al. Defective AMH signaling disrupts GnRH neuron development and function and contributes to hypogonadotropic hypogonadism. eLife 2019, 8, 10. [Google Scholar] [CrossRef]
- Cimino, I.; Casoni, F.; Liu, X.; Messina, A.; Parkash, J.; Jamin, S.P.; Catteau-Jonard, S.; Collier, F.; Baroncini, M.; Dewailly, D.; et al. Novel role for anti-Mullerian hormone in the regulation of GnRH neuron excitability and hormone secretion. Nat. Commun. 2016, 7, 10055. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuckey, B.G.A.; Jones, T.W.; Ward, B.K.; Wilson, S.G. Digenic Congenital Hypogonadotropic Hypogonadism Due to Heterozygous GNRH1 p.R31C and AMHR2 p.G445_L453del Variants. Genes 2023, 14, 1204. https://doi.org/10.3390/genes14061204
Stuckey BGA, Jones TW, Ward BK, Wilson SG. Digenic Congenital Hypogonadotropic Hypogonadism Due to Heterozygous GNRH1 p.R31C and AMHR2 p.G445_L453del Variants. Genes. 2023; 14(6):1204. https://doi.org/10.3390/genes14061204
Chicago/Turabian StyleStuckey, Bronwyn G. A., Timothy W. Jones, Bryan K. Ward, and Scott G. Wilson. 2023. "Digenic Congenital Hypogonadotropic Hypogonadism Due to Heterozygous GNRH1 p.R31C and AMHR2 p.G445_L453del Variants" Genes 14, no. 6: 1204. https://doi.org/10.3390/genes14061204
APA StyleStuckey, B. G. A., Jones, T. W., Ward, B. K., & Wilson, S. G. (2023). Digenic Congenital Hypogonadotropic Hypogonadism Due to Heterozygous GNRH1 p.R31C and AMHR2 p.G445_L453del Variants. Genes, 14(6), 1204. https://doi.org/10.3390/genes14061204