
A Comparison of Non-Destructive Visceral Swab and Tissue Biopsy Sampling Methods for Genotyping-by-Sequencing in the Freshwater Mussel Fusconaia askewi

 ,
, 
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Quantification, Library Preparation, and De Novo Assemblies
2.3. Number of SNPs and Coverage
2.4. Estimation of Genotype Probabilities
2.5. Data Analysis
3. Results
3.1. DNA Concentrations
3.2. SNP Numbers, Coverage, and Correlations
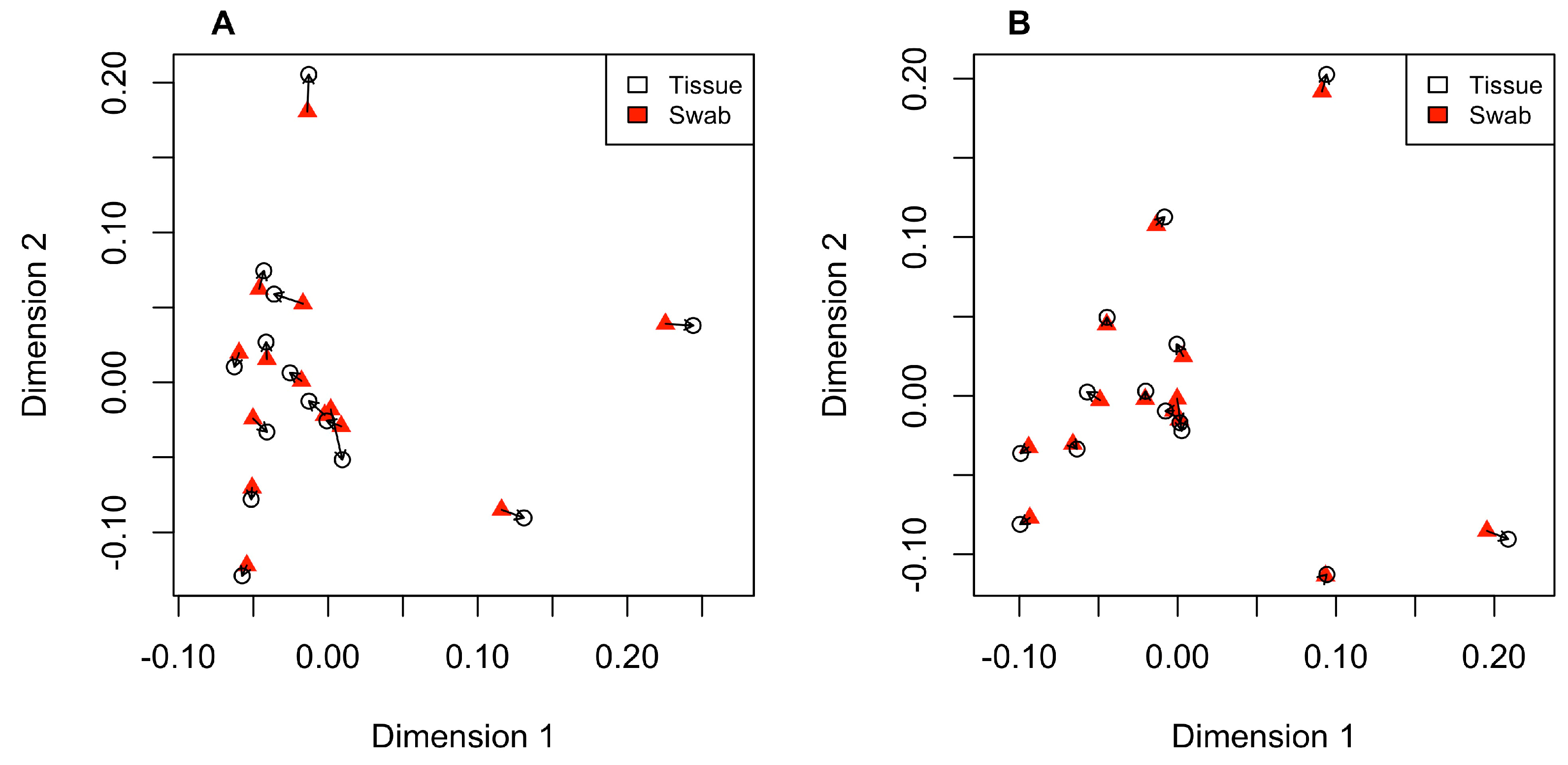
3.3. Comparing PCA Plots
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galbraith, H.S.; Zanatta, D.T.; Wilson, C.C. Comparative analysis of riverscape genetic structure in rare, threatened and common freshwater mussels. Conserv. Genet. 2015, 16, 845–857. [Google Scholar] [CrossRef]
- Kelly, M.W.; Rhymer, J.M. Population genetic structure of a rare unionid (Lampsilis cariosa) in a recently glaciated landscape. Conserv. Genet. 2005, 6, 789–802. [Google Scholar] [CrossRef]
- Lucas, L.K.; Fries, J.N.; Gabor, C.R.; Nice, C.C. Genetic variation and structure in Eurycea nana, a federally threatened salamander endemic to the San Marcos springs. J. Herpetol. 2009, 43, 220–227. [Google Scholar] [CrossRef]
- Nice, C.C.; Fordyce, J.A.; Sotola, V.A.; Crow, J.; Diaz, P.H. Geographic patterns of genomic variation in the threatened Salado salamander, Eurycea chisholmensis. Conserv. Genet. 2021, 22, 811–821. [Google Scholar] [CrossRef]
- Sotola, V.A.; Ruppel, D.S.; Bonner, T.H.; Nice, C.C.; Martin, N.H. Asymmetric introgression between fishes in the Red River basin of Texas is associated with variation in water quality. Ecol. Evol. 2019, 9, 2083–2095. [Google Scholar] [CrossRef]
- Banks, S.C.; Piggott, M.P. Non-invasive genetic sampling is one of our most powerful and ethical tools for threatened species population monitoring: A reply to Lavery et al. Biodivers. Conserv. 2022, 31, 723–728. [Google Scholar] [CrossRef]
- Baus, I.; Miño, C.I.; Monge, O. Current trends in the application of non-invasive genetic sampling to study Neotropical birds: Uses, goals, and conservation potential. Avian. Biol. Res. 2019, 12, 125–132. [Google Scholar] [CrossRef]
- Carroll, E.L.; Bruford, M.W.; DeWoody, J.A.; Leroy, G.; Strand, A.; Waits, L.; Wang, J. Genetic and genomic monitoring with minimally invasive sampling methods. Evol. Appl. 2018, 11, 1094–1119. [Google Scholar] [CrossRef]
- Storer, C.; Daniels, J.; Xiao, L.; Rossetti, K. Using noninvasive genetic sampling to survey rare butterfly populations. Insects 2019, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Waits, L.P.; Paetkau, D. Noninvasive genetic sampling tools for wildlife biologists: A review of applications and recommendations for accurate data collection. J. Wildl. Manag. 2005, 69, 1419–1433. [Google Scholar] [CrossRef]
- Graf, D.L.; Cummings, K.S. A ‘big data’ approach to global freshwater mussel diversity (Bivalvia: Unionoida), with an updated checklist of genera and species. J. Molluscan Stud. 2021, 87, eyaa034. [Google Scholar] [CrossRef]
- Lydeard, C.; Cowie, R.H.; Ponder, W.F.; Bogan, A.E.; Bouchet, P.; Clark, S.A.; Cummings, K.S.; Frest, T.J.; Gargominy, O.; Herbert, D.G.; et al. The global decline of nonmarine mollusks. BioScience 2004, 54, 321–330. [Google Scholar] [CrossRef]
- Williams, J.D.; Warren, M.L., Jr.; Cummings, K.S.; Harris, J.L.; Neves, R.J. Conservation status of freshwater mussels of the United States and Canada. Fisheries 1993, 18, 6–22. [Google Scholar] [CrossRef]
- Berg, D.J.; Haag, W.R.; Guttman, S.I.; Sickel, J.B. Mantle biopsy: A technique for nondestructive tissue-sampling of freshwater mussels. J. North Am. Benthol. Soc. 1995, 14, 577–581. [Google Scholar] [CrossRef]
- Henley, W.F.; Grobler, P.J.; Neves, R.J. Non-invasive method to obtain DNA from freshwater mussels (Bivalvia: Unionidae). J. Shellfish Res. 2006, 25, 975–977. [Google Scholar]
- Karlsson, S.; Larsen, B.M.; Eriksen, L.; Hagen, M. Four methods of nondestructive DNA sampling from freshwater pearl mussels Margaritifera margaritifera L.(Bivalvia: Unionoida). Freshw. Sci. 2013, 32, 525–530. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef]
- Massault, C.; Jeffrey, C.; Jones, D.B.; Barnard, R.; Strugnell, J.M.; Zenger, K.R.; Jerry, D.R. Non-invasive DNA collection for parentage analysis for bivalves: A case study from the silver-lipped pearl oyster (Pinctada maxima). Aquaculture 2022, 552, 738036. [Google Scholar] [CrossRef]
- Harrison, M. Population Genetic Structure of Unionid Mussels Across Multiple Gulf Drainages. Masters Thesis, Texas State University, San Marcos, TX, USA, 2022. [Google Scholar]
- Gompert, Z.; Lucas, L.K.; Nice, C.C.; Fordyce, J.A.; Forister, M.L.; Buerkle, C.A. Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution 2012, 66, 2167–2181. [Google Scholar] [CrossRef]
- Sung, C.J.; Bell, K.L.; Nice, C.C.; Martin, N.H. Integrating Bayesian genomic cline analyses and association mapping of morphological and ecological traits to dissect reproductive isolation and introgression in a Louisiana Iris hybrid zone. Mol. Ecol. 2018, 27, 959–978. [Google Scholar] [CrossRef]
- Zalmat, A.S.; Sotola, V.A.; Nice, C.C.; Martin, N.H. Genetic structure in Louisiana Iris species reveals patterns of recent and historical admixture. Am. J. Bot. 2021, 108, 2257–2268. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. Peer. J. 2014, 2, e431. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and com- paring large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Gompert, Z.; Lucas, L.K.; Buerkle, C.A.; Forister, M.L.; Fordyce, J.A.; Nice, C.C. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Mol. Ecol. 2014, 23, 4555–4573. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package, Version 2.5-6; 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 29 May 2023).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, M.; Sotola, V.A.; Zalmat, A.; Sullivan, K.T.; Littrell, B.M.; Bonner, T.H.; Martin, N.H. A Comparison of Non-Destructive Visceral Swab and Tissue Biopsy Sampling Methods for Genotyping-by-Sequencing in the Freshwater Mussel Fusconaia askewi. Genes 2023, 14, 1197. https://doi.org/10.3390/genes14061197
Harrison M, Sotola VA, Zalmat A, Sullivan KT, Littrell BM, Bonner TH, Martin NH. A Comparison of Non-Destructive Visceral Swab and Tissue Biopsy Sampling Methods for Genotyping-by-Sequencing in the Freshwater Mussel Fusconaia askewi. Genes. 2023; 14(6):1197. https://doi.org/10.3390/genes14061197
Chicago/Turabian StyleHarrison, Matthew, V. Alex Sotola, Alexander Zalmat, Kyle T. Sullivan, Bradley M. Littrell, Timothy H. Bonner, and Noland H. Martin. 2023. "A Comparison of Non-Destructive Visceral Swab and Tissue Biopsy Sampling Methods for Genotyping-by-Sequencing in the Freshwater Mussel Fusconaia askewi" Genes 14, no. 6: 1197. https://doi.org/10.3390/genes14061197
APA StyleHarrison, M., Sotola, V. A., Zalmat, A., Sullivan, K. T., Littrell, B. M., Bonner, T. H., & Martin, N. H. (2023). A Comparison of Non-Destructive Visceral Swab and Tissue Biopsy Sampling Methods for Genotyping-by-Sequencing in the Freshwater Mussel Fusconaia askewi. Genes, 14(6), 1197. https://doi.org/10.3390/genes14061197