
Identification of a Comprehensive Gene Co-Expression Network Associated with Autotetraploid Potato (Solanum tuberosum L.) Development Using WGCNA Analysis


Abstract
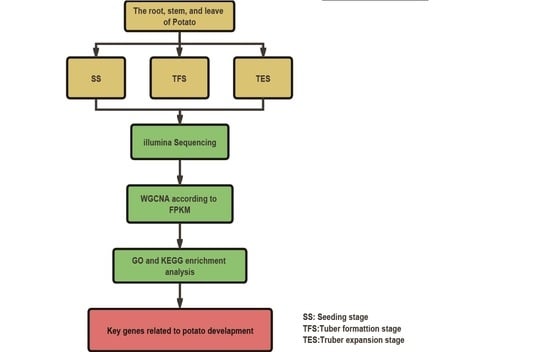
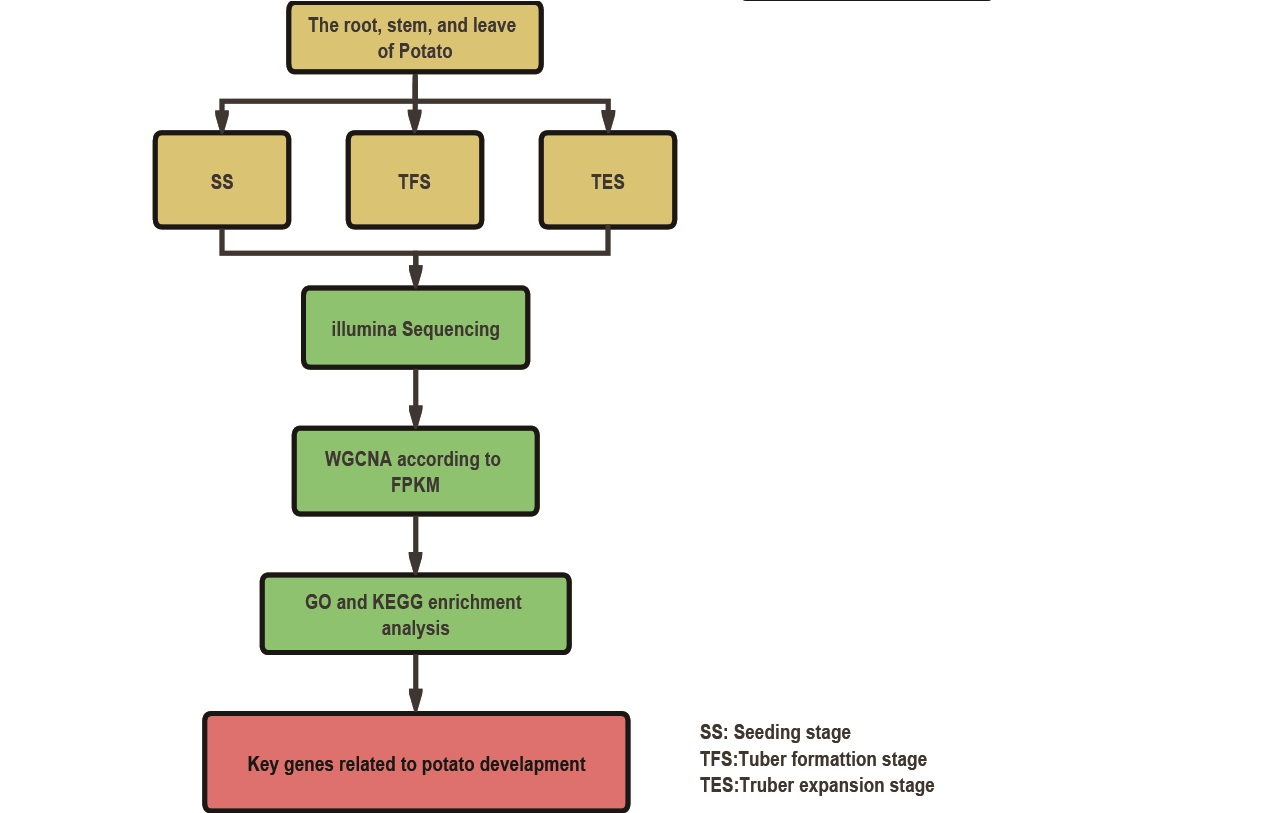{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Potato Culture and Sampling
2.2. RNA Extraction, Illumina Sequencing, and Data Analysis
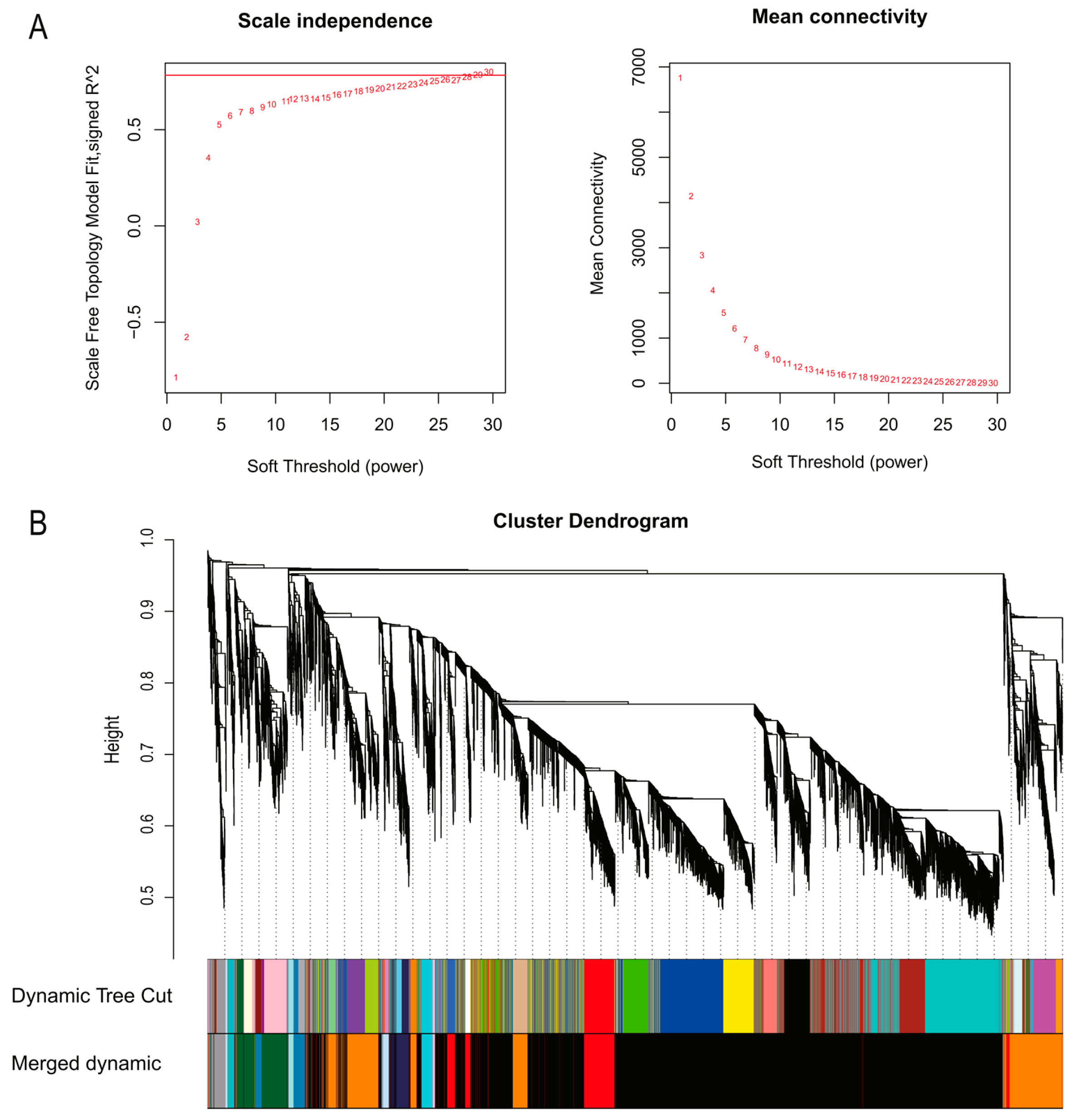
2.3. Weighted Co-Expressed Gene Network Construction
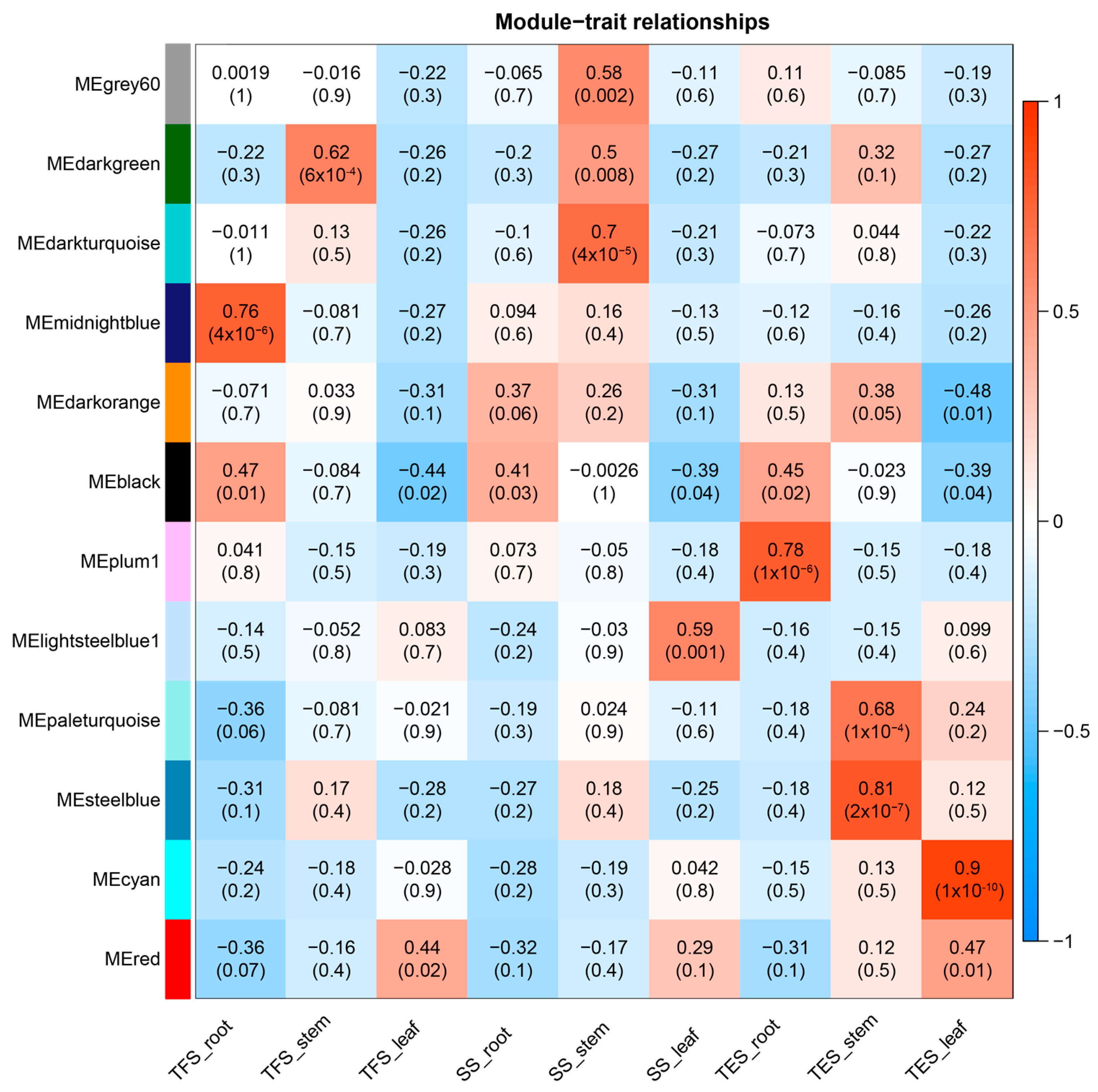
2.4. The Selection of Key Modules and Enrichment Analysis
2.5. Hub-Gene Screening and Functional Analysis
2.6. RT-qPCR Verification of Hub-Gene Expression
3. Results
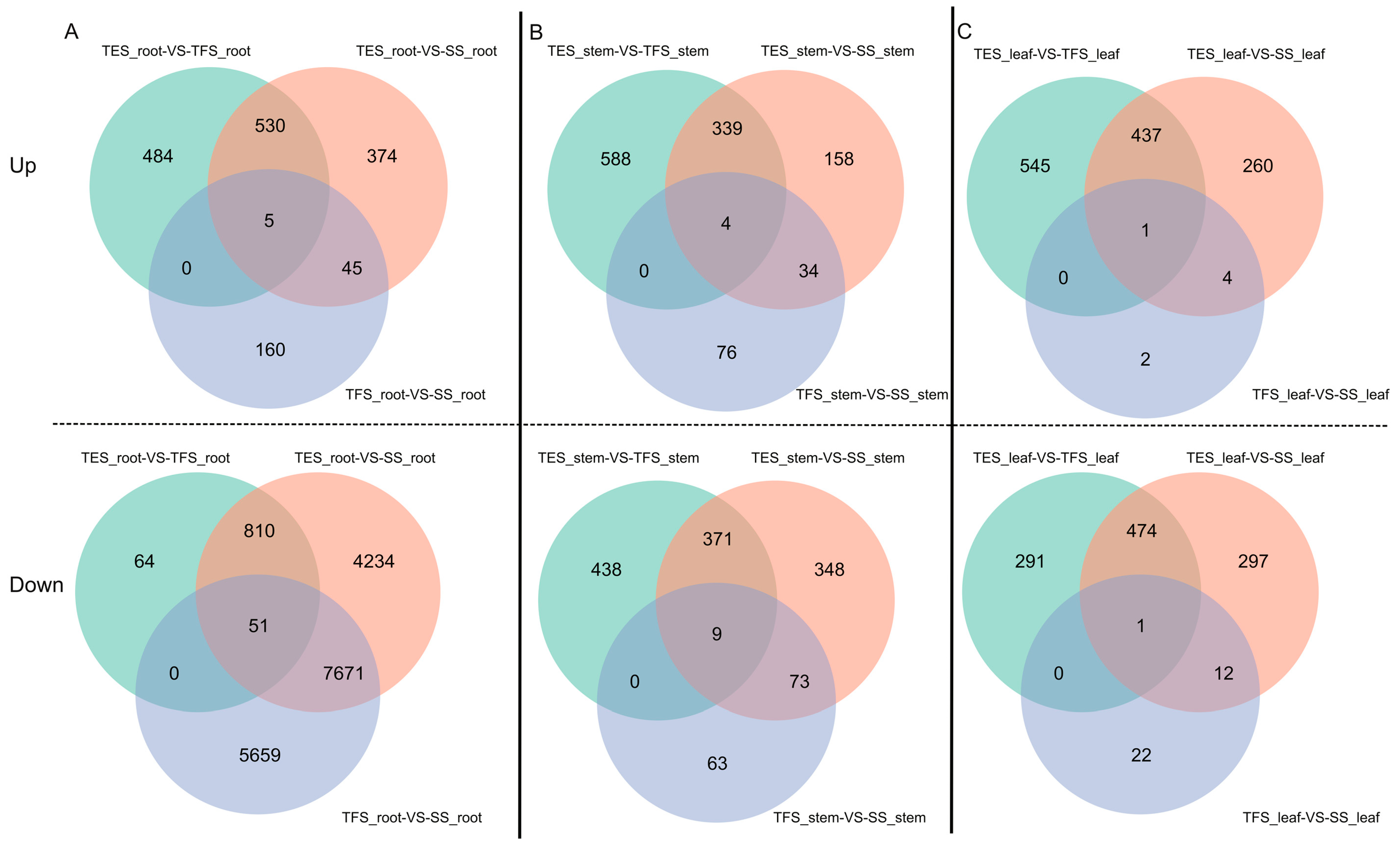
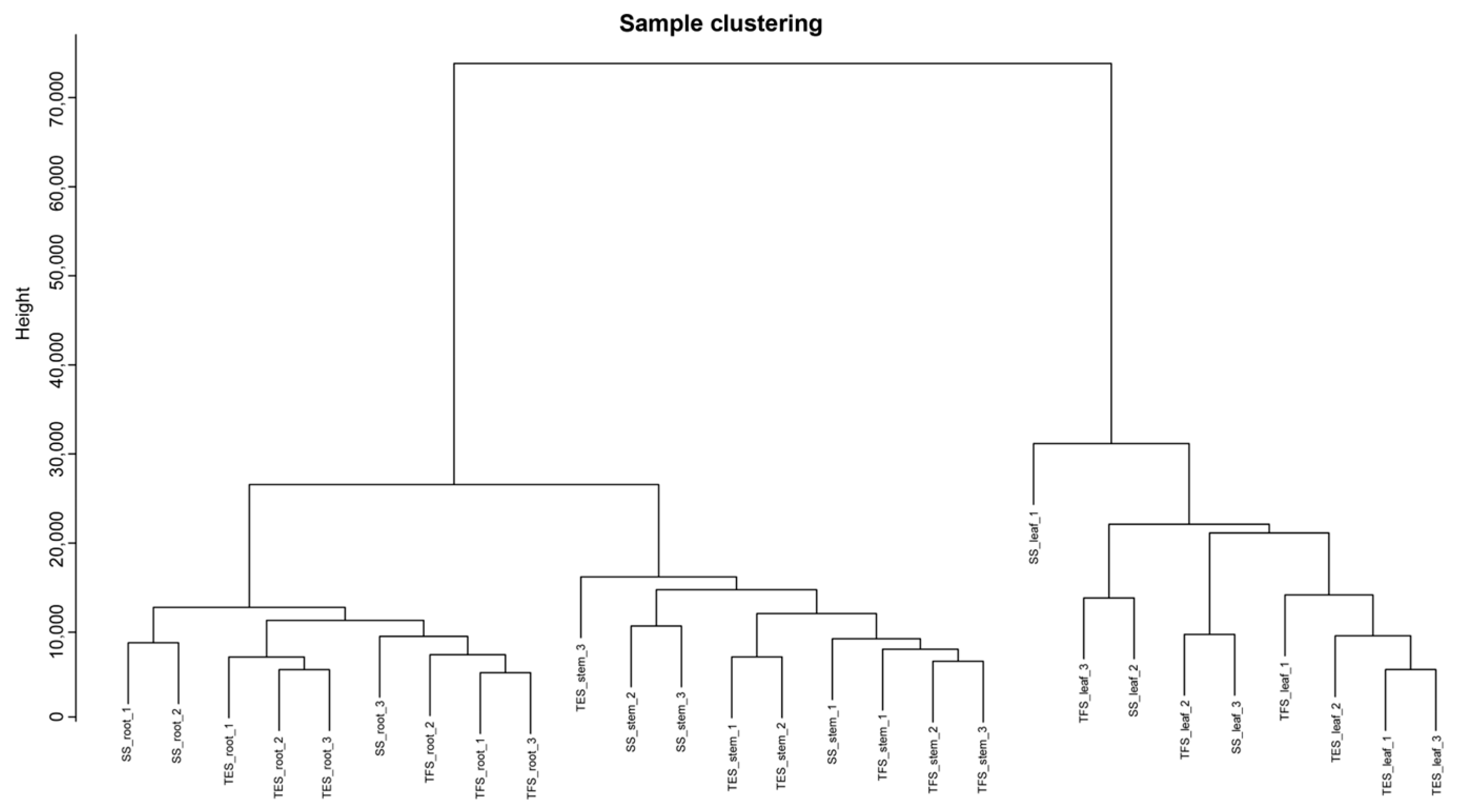
3.1. Genome-Wide Transcriptome Analysis
3.2. Weighted Gene Co-Expression Network Construction and Module Identification
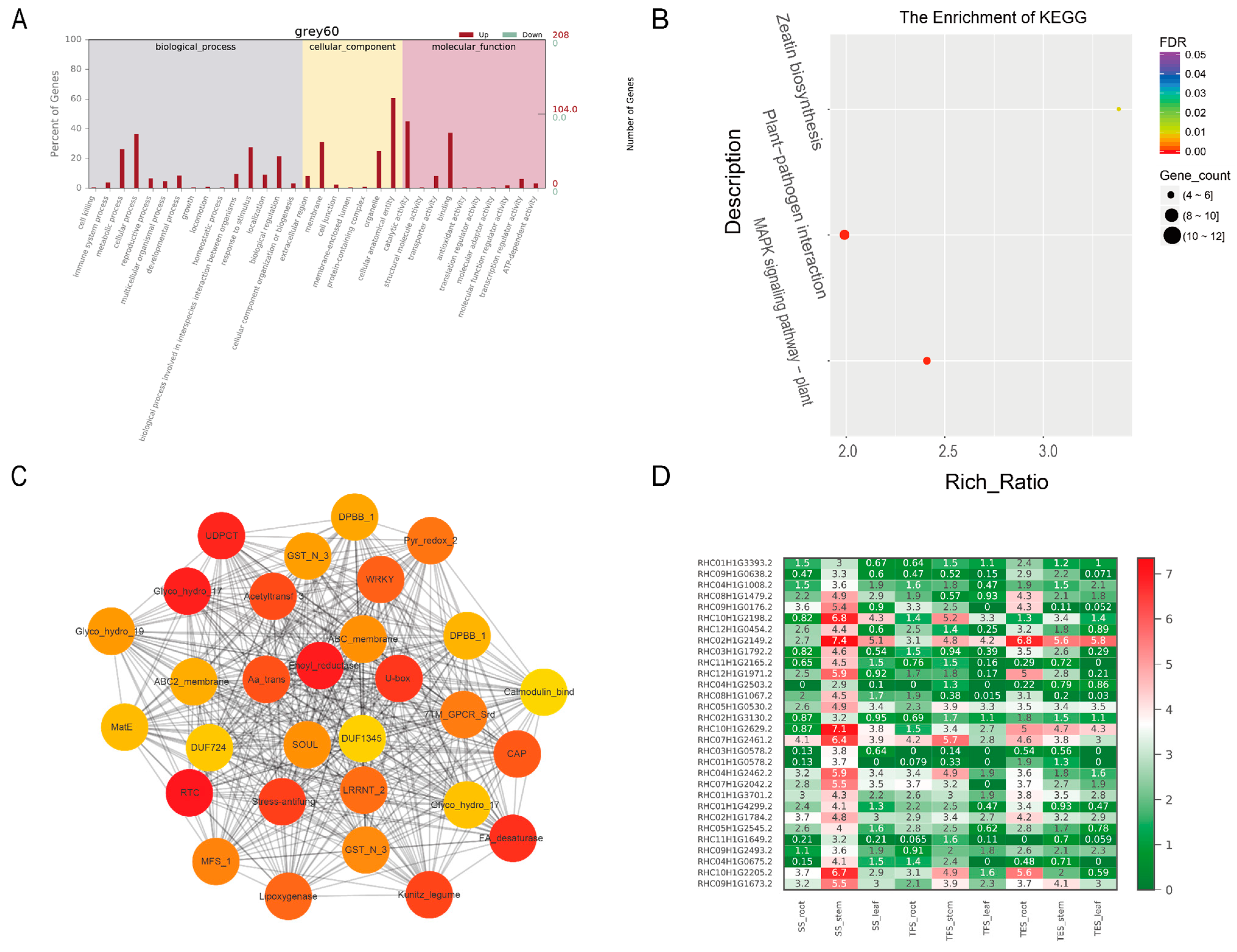
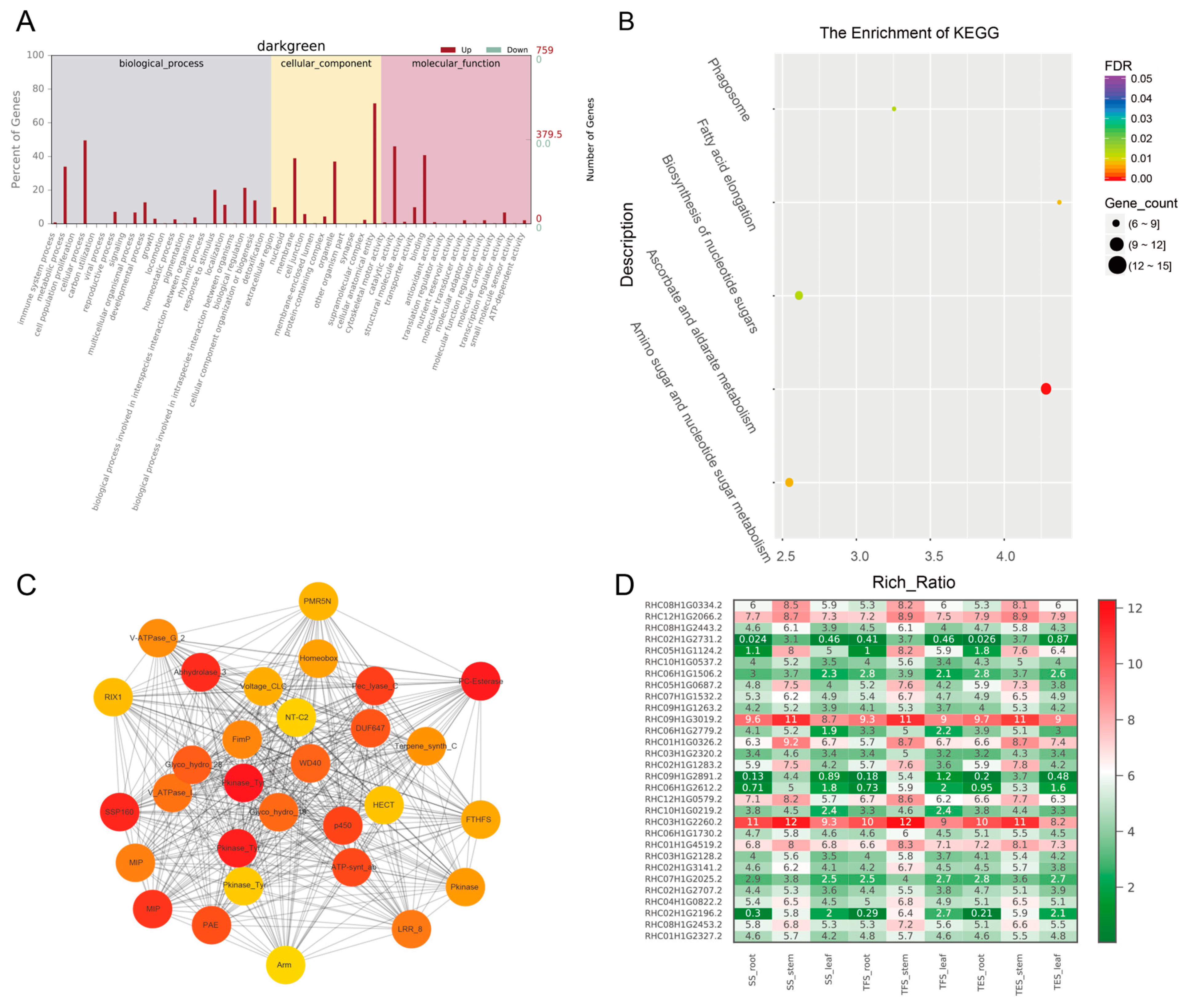
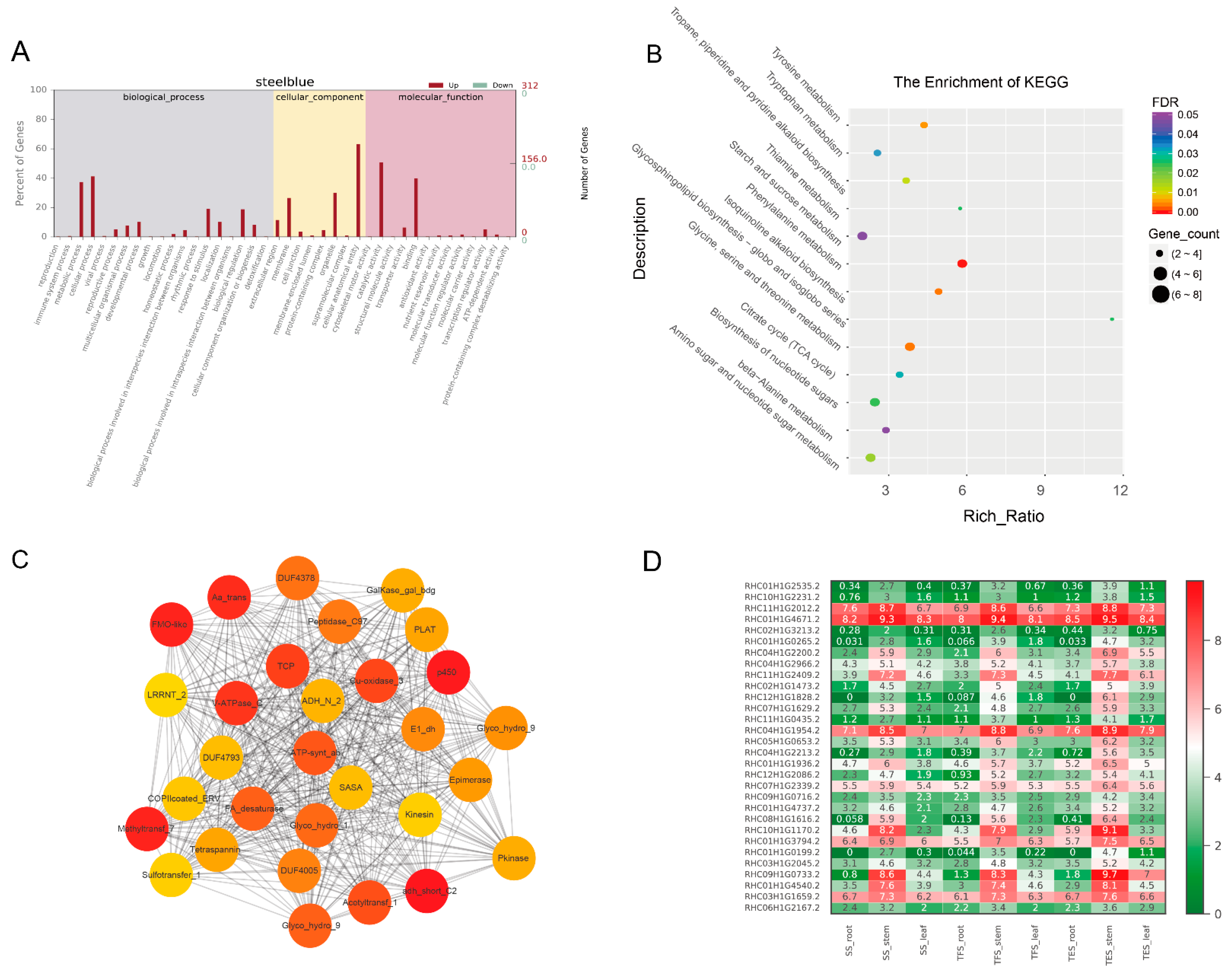
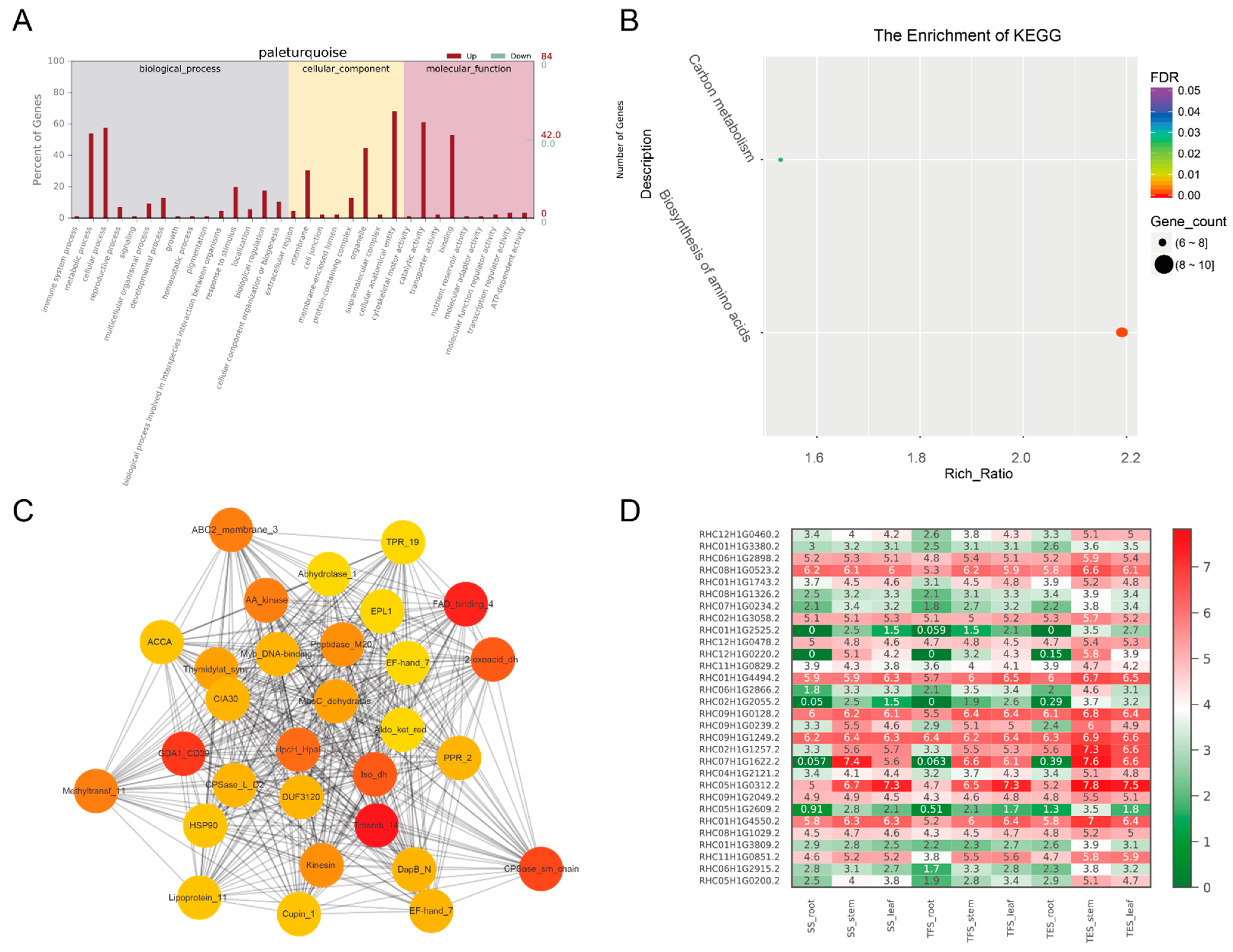
3.3. GO and KEGG Analysis of Key Modules in Different Tissues
3.4. Hub-Gene Screening and Functional Annotation
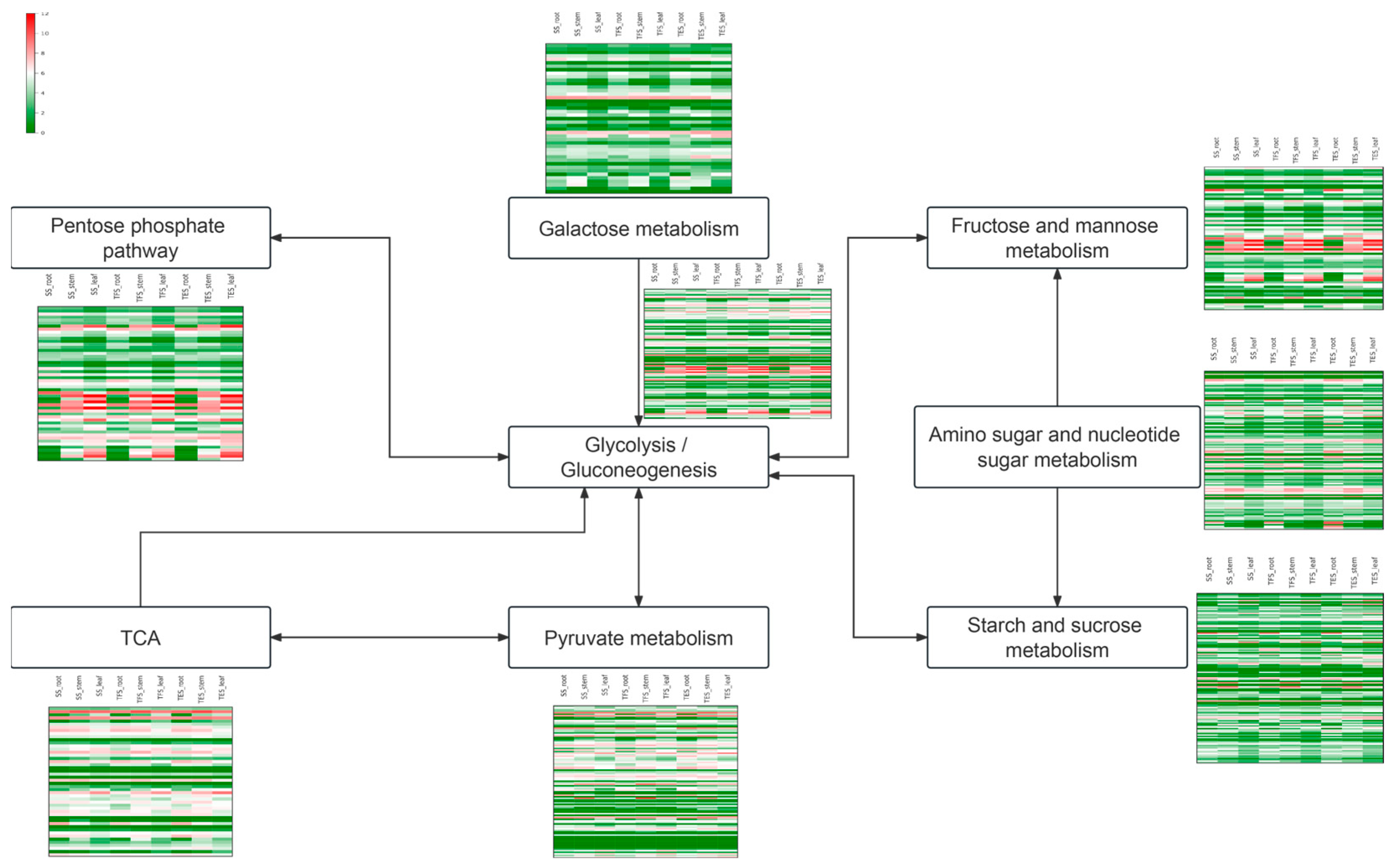
3.5. Changes in Gene Expression Levels of the Carbohydrate Metabolic Pathways
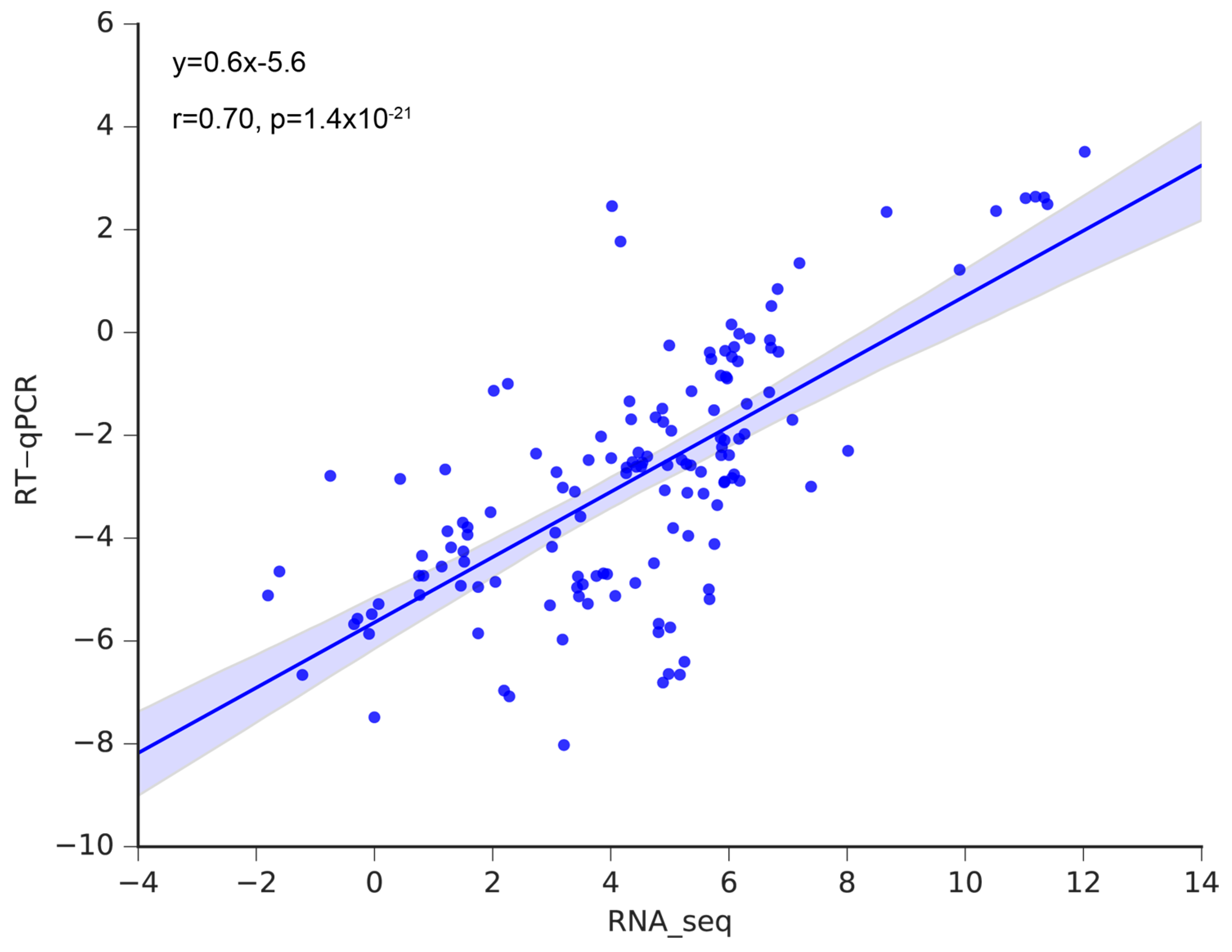
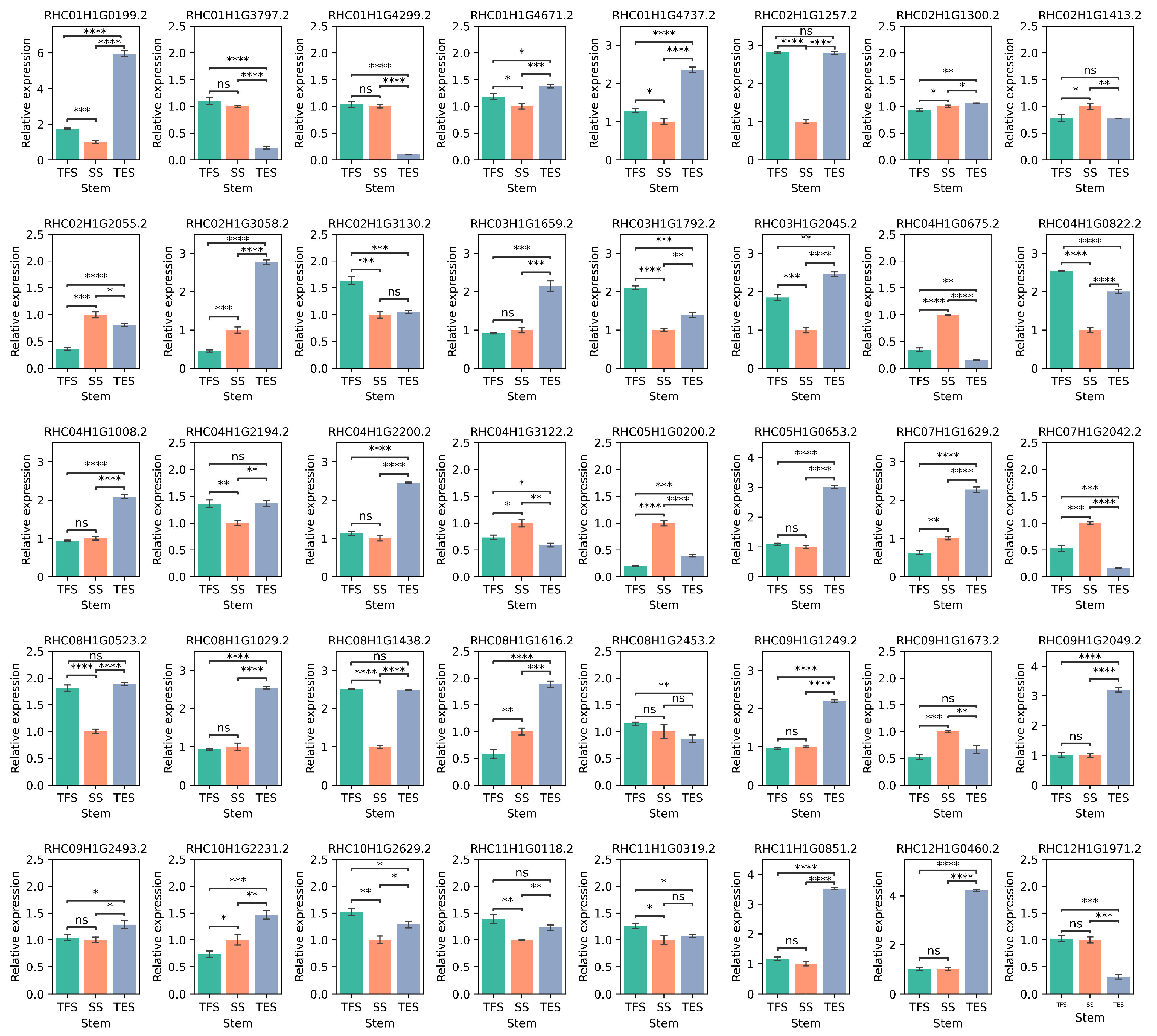
3.6. Validation of the Hub Genes Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Spooner, D.M.; McLean, K.; Ramsay, G.; Waugh, R.; Bryan, G.J. A single domestication for potato based on multilocus amplified fragment length polymorphism genotyping. Proc. Natl. Acad. Sci. USA 2005, 102, 14694–14699. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Huang, X.; Chen, J.; Luo, J.; Liu, Y.; Tang, Y.; Xiong, M.; Lu, Y.; Huang, Y.; Ouyang, B. Systematic Analysis of the Grafting-Related Glucanase-Encoding GH9 Family Genes in Pepper, Tomato and Tobacco. Plants 2022, 11, 2092. [Google Scholar] [CrossRef]
- Yan, W.; Jian, Y.; Duan, S.; Guo, X.; Hu, J.; Yang, X.; Li, G. Dissection of the Plant Hormone Signal Transduction Network in Late Blight Resistant Potato Genotype SD20 and Prediction of Key Resistance Genes. Phytopathology 2022, 113, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Hou, H.; Meng, X.; Chen, A.; Wang, L.; Zhu, H.; Zheng, S.; Lv, Z.; Zhu, X. Transcriptome analysis reveals the proline metabolic pathway and its potential regulation TF-hub genes in salt-stressed potato. Front. Plant Sci. 2022, 13, 1030138. [Google Scholar] [CrossRef]
- Guo, H.; Pu, X.; Jia, H.; Zhou, Y.; Ye, G.; Yang, Y.; Na, T.; Wang, J. Transcriptome analysis reveals multiple effects of nitrogen accumulation and metabolism in the roots, shoots, and leaves of potato (Solanum tuberosum L.). BMC Plant Biol. 2022, 22, 282. [Google Scholar] [CrossRef]
- Cao, W.; Gan, L.; Shang, K.; Wang, C.; Song, Y.; Liu, H.; Zhou, S.; Zhu, C. Global transcriptome analyses reveal the molecular signatures in the early response of potato (Solanum tuberosum L.) to Phytophthora infestans, Ralstonia solanacearum, and Potato virus Y infection. Planta 2020, 252, 57. [Google Scholar] [CrossRef]
- Liu, M.; Bassetti, N.; Petrasch, S.; Zhang, N.; Bucher, J.; Shen, S.; Zhao, J.; Bonnema, G. What makes turnips: Anatomy, physiology and transcriptome during early stages of its hypocotyl-tuber development. Hortic. Res. 2019, 6, 38. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sun, H.; Jiao, W.B.; Krause, K.; Campoy, J.A.; Goel, M.; Folz-Donahue, K.; Kukat, C.; Huettel, B.; Schneeberger, K. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 2022, 54, 342–348. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, F.; Chen, Y.; Fan, Y.; Zhang, K.; Liu, Q.; Tu, W.; Jiang, F.; Li, G.; Zhao, H.; et al. Salicylic Acid Improves the Constitutive Freezing Tolerance of Potato as Revealed by Transcriptomics and Metabolomics Analyses. Int. J. Mol. Sci. 2022, 24, 609. [Google Scholar] [CrossRef]
- Glushkevich, A.; Spechenkova, N.; Fesenko, I.; Knyazev, A.; Samarskaya, V.; Kalinina, N.O.; Taliansky, M.; Love, A.J. Transcriptomic Reprogramming, Alternative Splicing and RNA Methylation in Potato (Solanum tuberosum L.) Plants in Response to Potato Virus Y Infection. Plants 2022, 11, 635. [Google Scholar] [CrossRef]
- Qaseem, M.F.; Wu, A.M. Balanced Xylan Acetylation is the Key Regulator of Plant Growth and Development, and Cell Wall Structure and for Industrial Utilization. Int. J. Mol. Sci. 2020, 21, 7875. [Google Scholar] [CrossRef]
- Si, C.; Teixeira da Silva, J.A.; He, C.; Yu, Z.; Zhao, C.; Wang, H.; Zhang, M.; Duan, J. DoRWA3 from Dendrobium officinale Plays an Essential Role in Acetylation of Polysaccharides. Int. J. Mol. Sci. 2020, 21, 6250. [Google Scholar] [CrossRef]
- Datir, S.; Regan, S. Advances in Physiological, Transcriptomic, Proteomic, Metabolomic, and Molecular Genetic Approaches for Enhancing Mango Fruit Quality. J. Agric. Food Chem. 2023, 71, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, C.; Ric-Varas, P.; Garcia-Gago, J.A.; Lopez-Casado, G.; Blanco-Portales, R.; Munoz-Blanco, J.; Schuckel, J.; Knox, J.P.; Matas, A.J.; Quesada, M.A.; et al. Elucidating the role of polygalacturonase genes in strawberry fruit softening. J. Exp. Bot. 2020, 71, 7103–7117. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yi, F.; Ling, J.; Wang, Z.; Zhao, K.; Lu, N.; Qu, G.; Kong, L.; Ma, W.; Wang, J. Transcriptomics and Proteomics Reveal the Cellulose and Pectin Metabolic Processes in the Tension Wood (Non-G-Layer) of Catalpa bungee. Int. J. Mol. Sci. 2020, 21, 1686. [Google Scholar] [CrossRef]
- Zaveska Drabkova, L.; Honys, D.; Motyka, V. Evolutionary diversification of cytokinin-specific glucosyltransferases in angiosperms and enigma of missing cis-zeatin O-glucosyltransferase gene in Brassicaceae. Sci. Rep. 2021, 11, 7885. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, K.; Zhang, Z.; Liu, Y.; Lv, F.; Sun, P.; Gao, S.; Wang, Q.; Yu, C.; Jiang, J.; et al. A chromosome-level genome assembly for Dracaena cochinchinensis reveals the molecular basis of its longevity and formation of dragon’s blood. Plant Commun. 2022, 3, 100456. [Google Scholar] [CrossRef]
- Bueno, T.V.; Fontes, P.P.; Abe, V.Y.; Utiyama, A.S.; Senra, R.L.; Oliveira, L.S.; Brombini Dos Santos, A.; Ferreira, E.G.C.; Darben, L.M.; de Oliveira, A.B.; et al. A Phakopsora pachyrhizi Effector Suppresses PAMP-Triggered Immunity and Interacts with a Soybean Glucan Endo-1,3-beta-Glucosidase to Promote Virulence. Mol. Plant Microbe. Interact. 2022, 35, 779–790. [Google Scholar] [CrossRef]
- Yu, M.; Liu, H.; Guo, L.; Zhou, T.; Shan, Y.; Xia, Z.; Li, X.; An, M.; Wu, Y. Antiviral modes of action of the novel compound GLY-15 containing pyrimidine heterocycle and moroxydine skeleton against tobacco mosaic virus. Pest. Manag. Sci. 2022, 78, 5259–5270. [Google Scholar] [CrossRef]
- Singh, L.; Coronejo, S.; Pruthi, R.; Chapagain, S.; Subudhi, P.K. Integration of QTL Mapping and Whole Genome Sequencing Identifies Candidate Genes for Alkalinity Tolerance in Rice (Oryza sativa). Int. J. Mol. Sci. 2022, 23, 11791. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.Q.; Zou, Y.; Zheng, Q.W.; Liu, Y.L.; Li, R.R.; Lin, J.F.; Guo, L.Q. TMT-MS/MS proteomic analysis of the carbohydrate-active enzymes in the fruiting body of Pleurotus tuoliensis during storage. J. Sci. Food Agric. 2021, 101, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, L.; Sun, L.; Lin, B.; Huang, K.; Zhuo, K.; Liao, J. A novel Meloidogyne graminicola effector, MgMO237, interacts with multiple host defence-related proteins to manipulate plant basal immunity and promote parasitism. Mol. Plant Pathol. 2018, 19, 1942–1955. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yang, X.; Feng, M.; Ding, H.; Khan, M.T.; Zhang, J.; Zhang, M. Genome-wide analysis of R2R3-MYB transcription factors family in the autopolyploid Saccharum spontaneum: An exploration of dominance expression and stress response. BMC Genom. 2021, 22, 622. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, J.; Lang, X.; Han, D.; Hu, Y.; Ding, Y.; Wang, G.; Guo, J.; Shi, L. Integrating transcriptomic and metabolomic analysis in roots of wild soybean seedlings in response to low-phosphorus stress. Front. Plant Sci. 2022, 13, 1006806. [Google Scholar] [CrossRef]
- Lv, Y.; Ma, J.; Wang, Y.; Wang, Q.; Lu, X.; Hu, H.; Qian, Q.; Guo, L.; Shang, L. Loci and Natural Alleles for Low-Nitrogen-Induced Growth Response Revealed by the Genome-Wide Association Study Analysis in Rice (Oryza sativa L.). Front. Plant Sci. 2021, 12, 770736. [Google Scholar] [CrossRef]
- Yang, T.; Li, C.; Shen, J.; Zhuang, B.; Wen, Y. [Genome-wide identification and analysis of the TCP gene family in eggplant (Solanum melongena L.)]. Sheng Wu Gong Cheng Xue Bao 2022, 38, 2974–2988. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Q.; Zheng, J.; Xu, F.; Ye, J.; Zhang, W.; Liao, Y.; Yang, X. Genome-wide identification and expression pattern analysis of the TCP transcription factor family in Ginkgo biloba. Plant Signal. Behav. 2022, 17, 1994248. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, J.; Wang, J. Identification of a Comprehensive Gene Co-Expression Network Associated with Autotetraploid Potato (Solanum tuberosum L.) Development Using WGCNA Analysis. Genes 2023, 14, 1162. https://doi.org/10.3390/genes14061162
Li Z, Wang J, Wang J. Identification of a Comprehensive Gene Co-Expression Network Associated with Autotetraploid Potato (Solanum tuberosum L.) Development Using WGCNA Analysis. Genes. 2023; 14(6):1162. https://doi.org/10.3390/genes14061162
Chicago/Turabian StyleLi, Zhimin, Juan Wang, and Jiayin Wang. 2023. "Identification of a Comprehensive Gene Co-Expression Network Associated with Autotetraploid Potato (Solanum tuberosum L.) Development Using WGCNA Analysis" Genes 14, no. 6: 1162. https://doi.org/10.3390/genes14061162
APA StyleLi, Z., Wang, J., & Wang, J. (2023). Identification of a Comprehensive Gene Co-Expression Network Associated with Autotetraploid Potato (Solanum tuberosum L.) Development Using WGCNA Analysis. Genes, 14(6), 1162. https://doi.org/10.3390/genes14061162