
Evaluating the Usefulness of Human DNA Quantification to Predict DNA Profiling Success of Historical Bone Samples

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Skeletal Samples
2.2. Sample Preparation and DNA Extraction
2.3. DNA Quantification
2.4. Next Generation Sequencing
2.4.1. Library Preparation
2.4.2. Hybridization Capture
2.4.3. Normalization and Pooling
2.4.4. Loading and Sequencing
2.4.5. Sequence Data Analysis
2.5. STR Typing
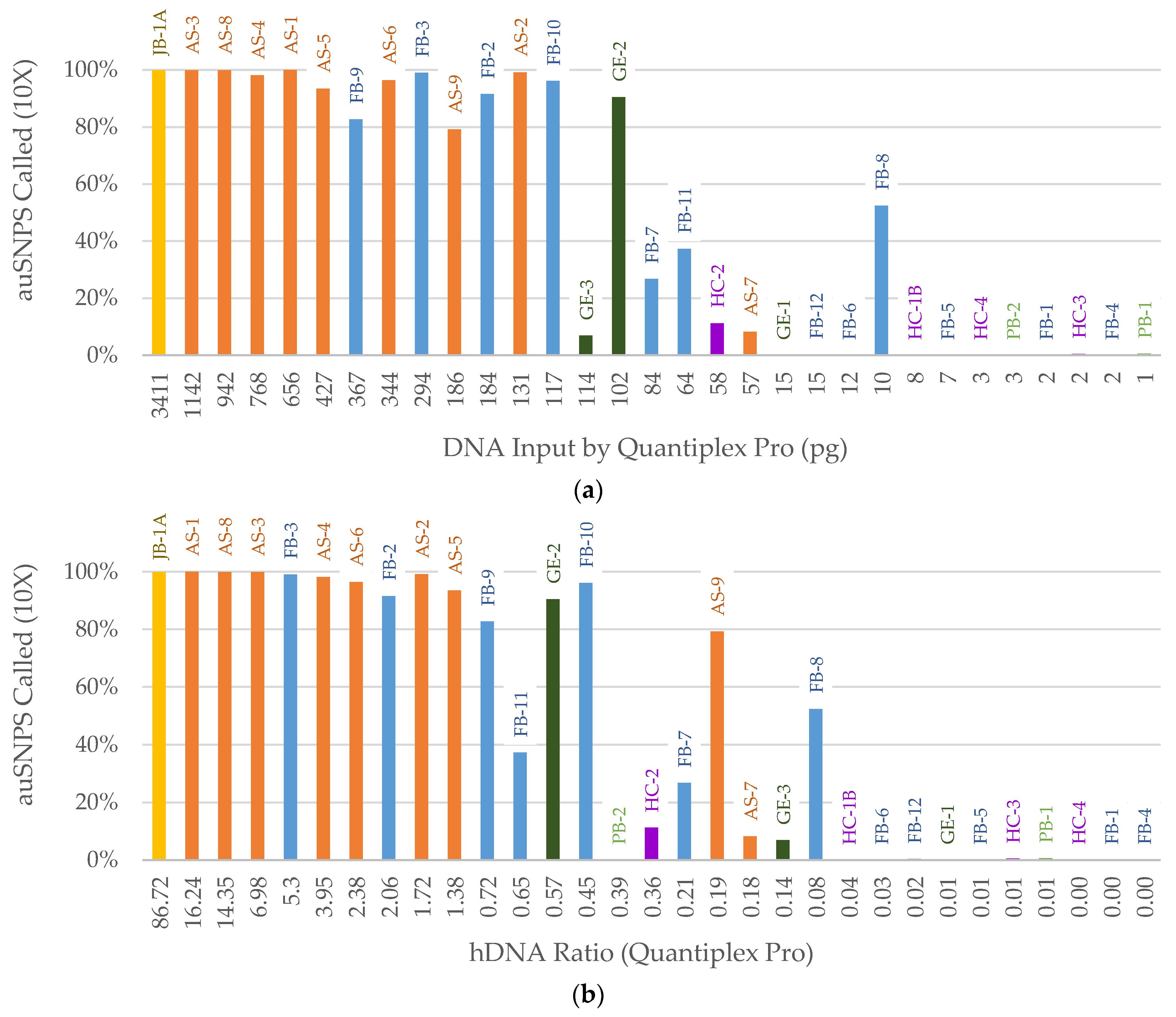
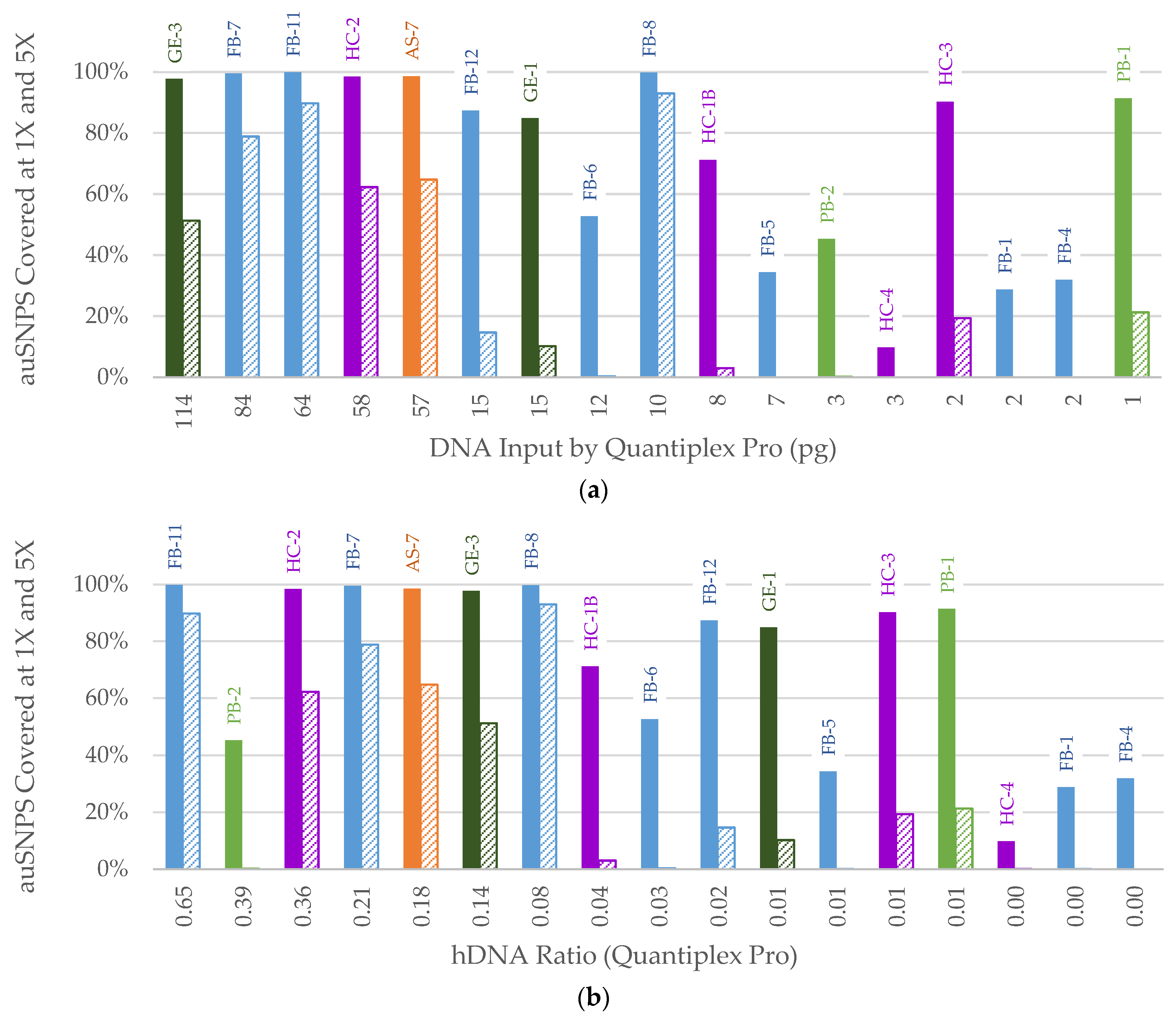
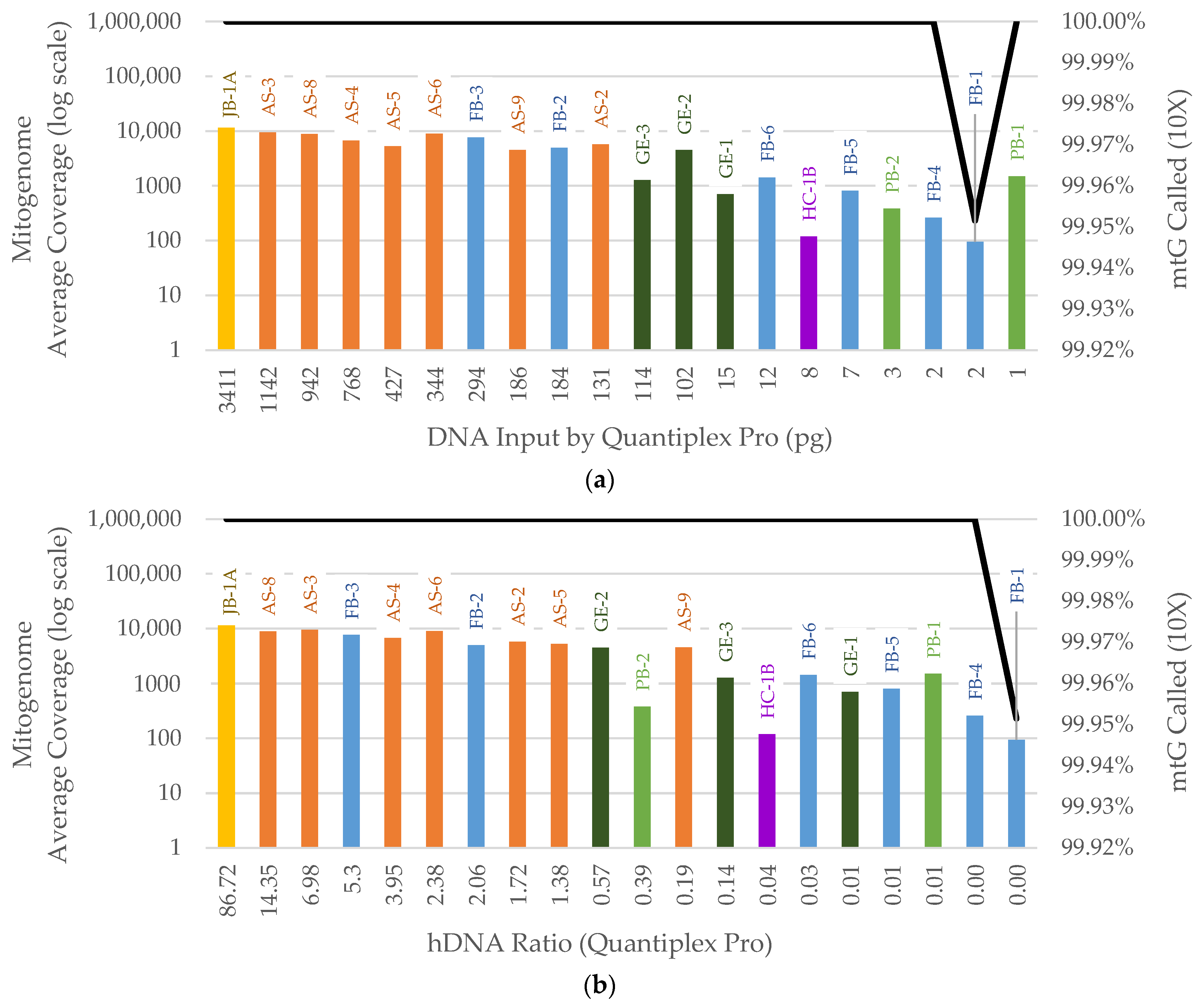
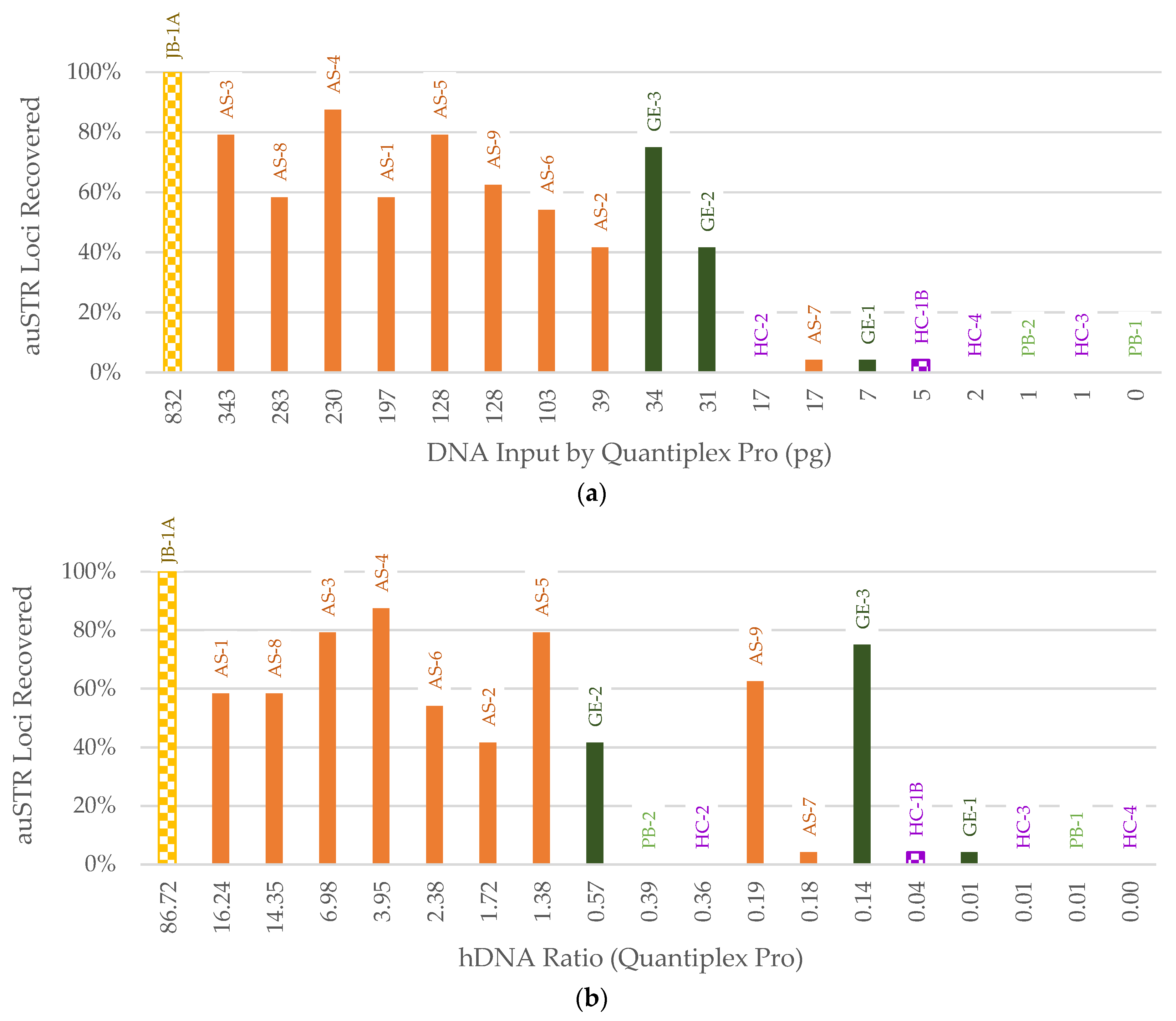
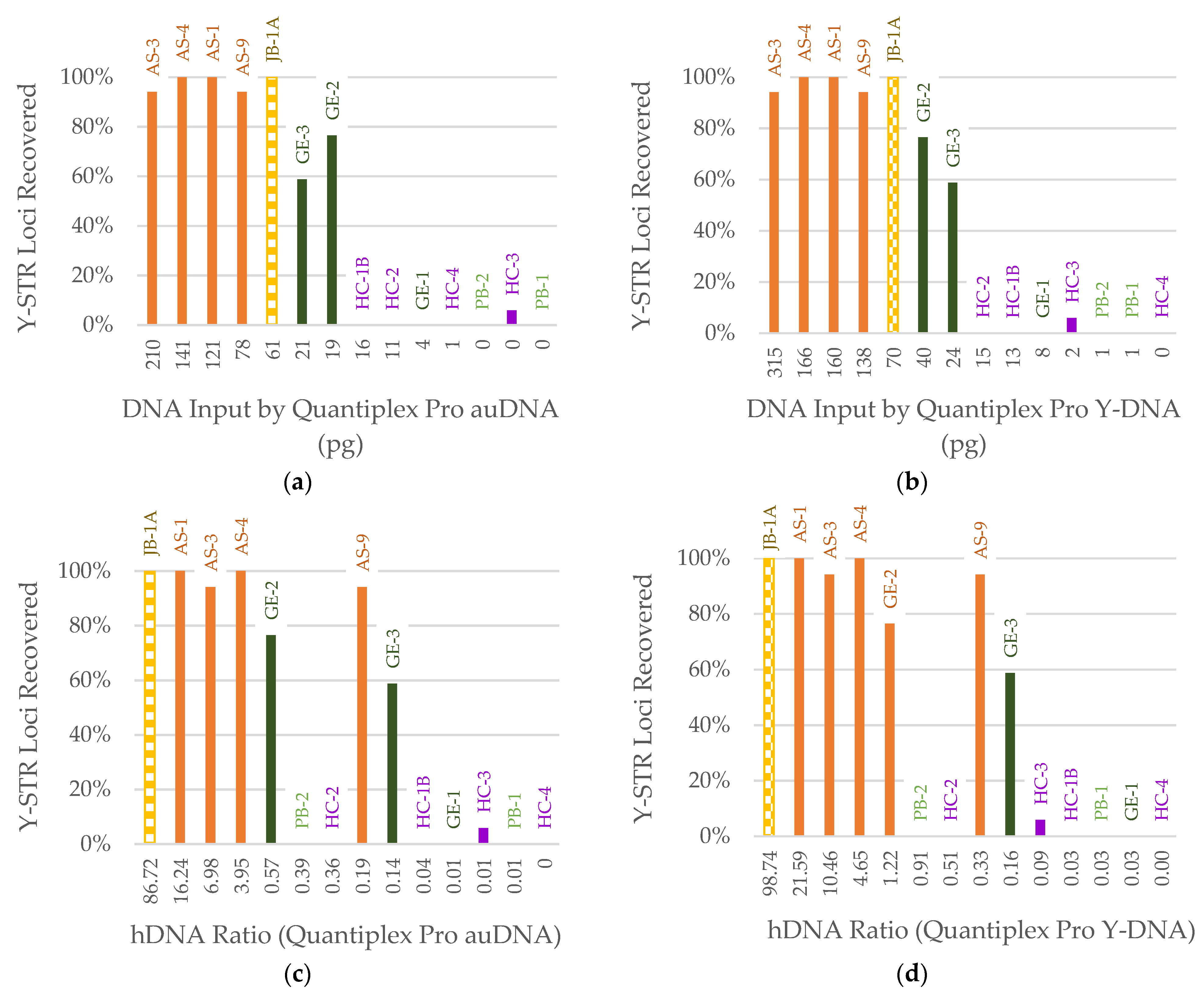
3. Results
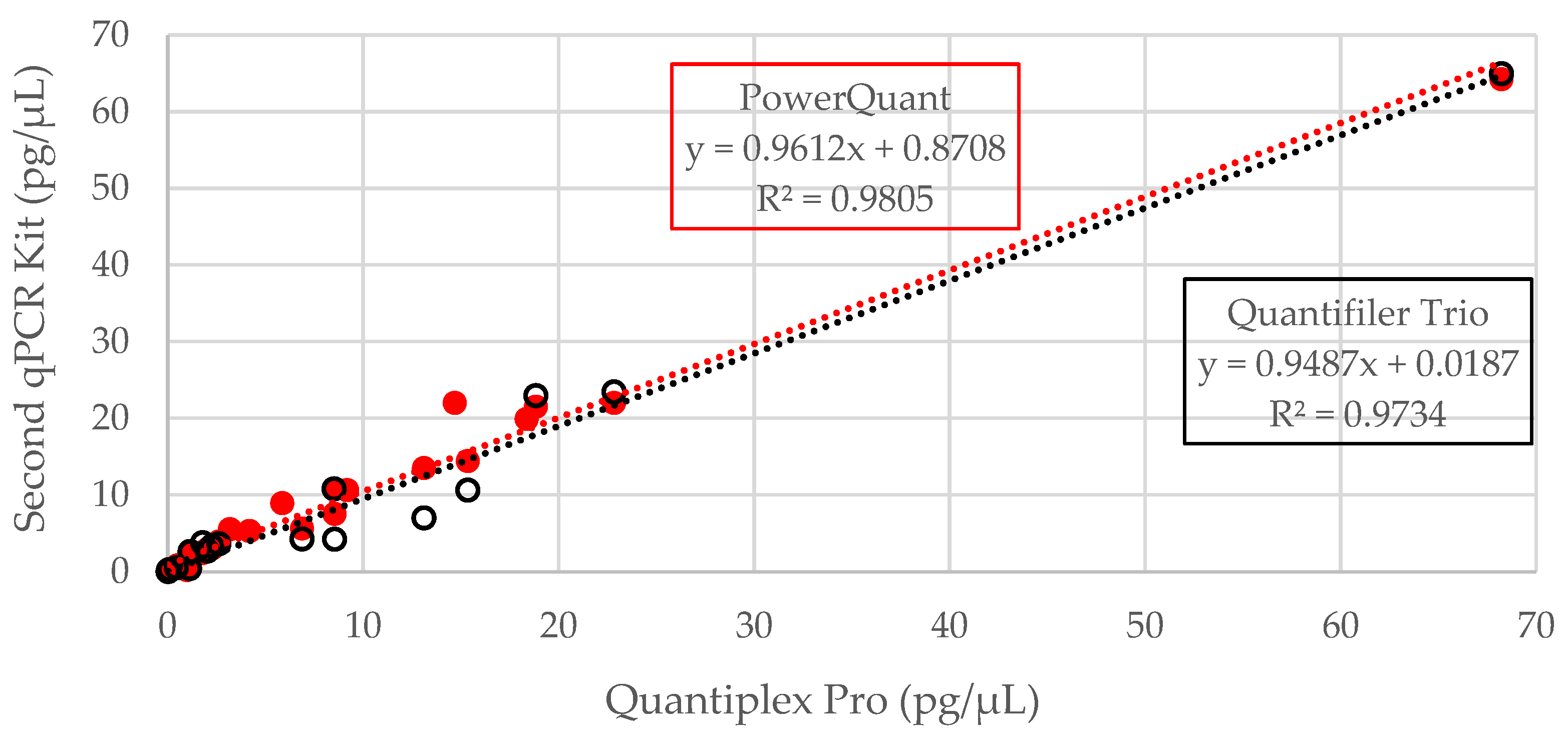
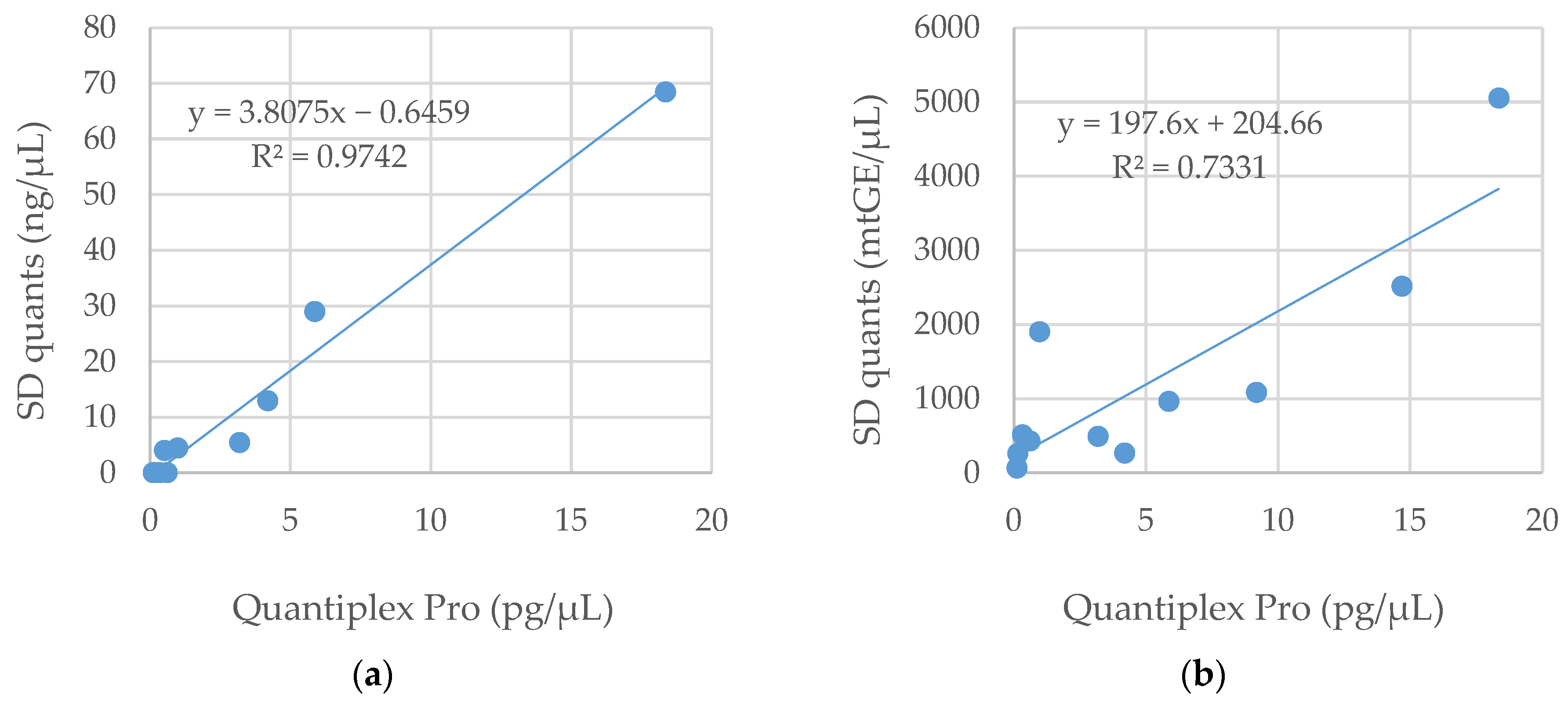
3.1. DNA Quantification
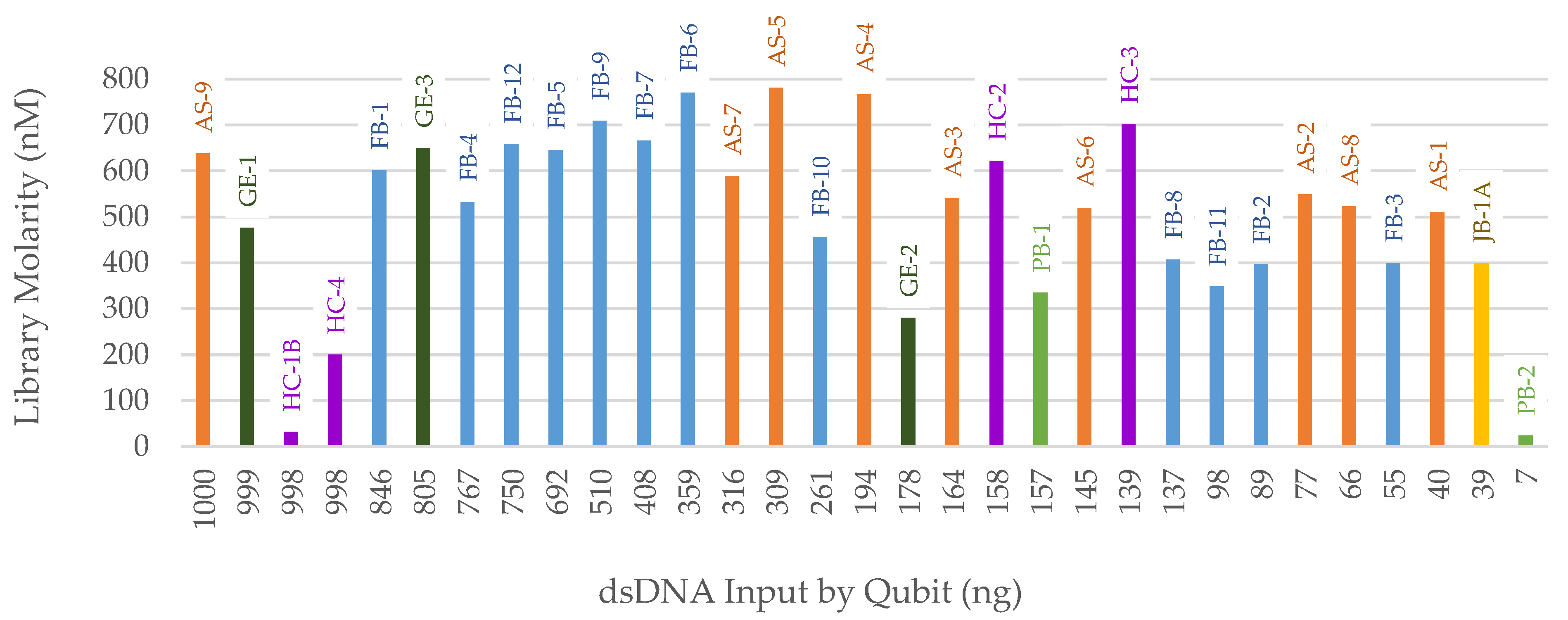
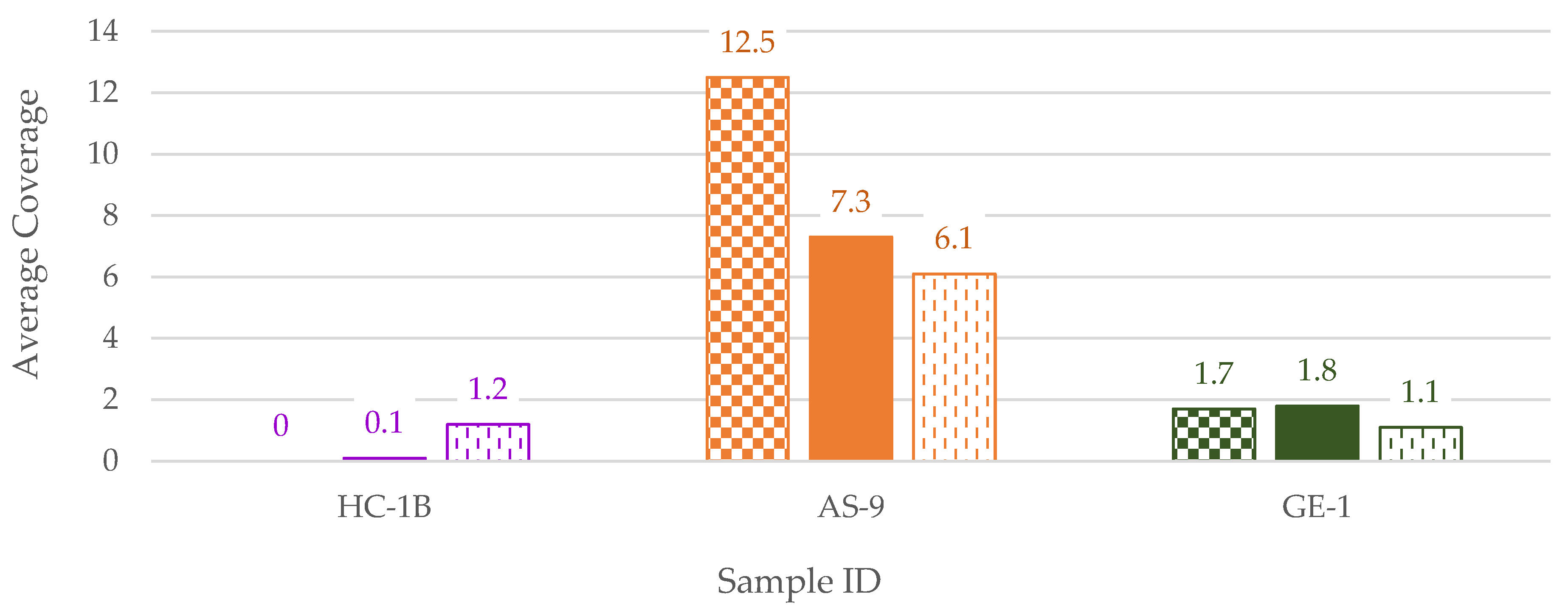
3.2. NGS
3.2.1. FORCE Capture
3.2.2. Mitogenome Capture
3.3. STR Typing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef] [PubMed]
- Zavala, E.I.; Thomas, J.T.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Meyers, K.K.; Barrit-Ross, S.; Aximu-Petri, A.; Richter, J.; Nickel, B.; Berg, G.E.; et al. Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns. Genes 2022, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Gorden, E.M.; Sturk-Andreaggi, K.; Warnke-Sommer, J.; Hazelwood, A.; Barritt-Ross, S.; Marshall, C. Next generation sequencing of STR artifacts produced from historical bone samples. Forensic Sci. Int. Genet. 2020, 49, 102397. [Google Scholar] [CrossRef] [PubMed]
- Finaughty, C.; Heathfield, L.J.; Kemp, V.; Márquez-Grant, N. Forensic DNA extraction methods for human hard tissue: A systematic literature review and meta-analysis of technologies and sample type. Forensic Sci. Int. Genet. 2022, 63, 102818. [Google Scholar] [CrossRef]
- Marshall, C.; Sturk-Andreaggi, K.; Gorden, E.M.; Daniels-Higginbotham, J.; Sanchez, S.G.; Bašić, Ž.; Kružić, I.; Anđelinović, Š.; Bosnar, A.; Čoklo, M.; et al. A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulić. Genes 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Kapp, J.D.; Green, R.E.; Shapiro, B. A Fast and Efficient Single-stranded Genomic Library Preparation Method Optimized for Ancient DNA. J. Hered. 2021, 112, 241–249. [Google Scholar] [CrossRef]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef]
- Enk, J.; Rouillard, J.-M.; Poinar, H. Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. Biotechniques 2013, 55, 300–309. [Google Scholar] [CrossRef]
- Brzobohatá, K.; Drozdová, E.; Smutný, J.; Zeman, T.; Beňuš, R. Comparison of Suitability of the Most Common Ancient DNA Quantification Methods. Genet. Test. Mol. Biomark. 2017, 21, 265–271. [Google Scholar] [CrossRef]
- Xavier, C.; Eduardoff, M.; Strobl, C.; Parson, W. SD quants—Sensitive detection tetraplex-system for nuclear and mitochondrial DNA quantification and degradation inference. Forensic Sci. Int. Genet. 2019, 42, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kavlick, M.F. Development of a triplex mtDNA qPCR assay to assess quantification, degradation, inhibition, and amplification target copy numbers. Mitochondrion 2018, 46, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Daniels-Higginbotham, J.; Gorden, E.M.; Farmer, S.K.; Spatola, B.; Damann, F.; Bellantoni, N.; Gagnon, K.S.; de la Puente, M.; Xavier, C.; Walsh, S.; et al. DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes 2019, 10, 636. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do It Right or Not at All. Science 2000, 289, 1139. [Google Scholar] [CrossRef]
- Hofreiter, M.; Sneberger, J.; Pospisek, M.; Vanek, D. Progress in forensic bone DNA analysis: Lessons learned from ancient DNA. Forensic Sci. Int. Genet. 2021, 54, 102538. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; Eduardoff, M.; Bertoglio, B.; Amory, C.; Berger, C.; Casas-Vargas, A.; Pallua, J.; Parson, W. Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age. Genes 2021, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Tillmar, A.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Thomas, J.T.; Marshall, C. The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes 2021, 12, 1968. [Google Scholar] [CrossRef]
- Gorden, E.M.; Sturk-Andreaggi, K.; Marshall, C. Repair of DNA damage caused by cytosine deamination in mitochondrial DNA of forensic case samples. Forensic Sci. Int. Genet. 2018, 34, 257–264. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Peck, M.A.; Boysen, C.; Dekker, P.; McMahon, T.P.; Marshall, C.K. AQME: A forensic mitochondrial DNA analysis tool for next-generation sequencing data. Forensic Sci. Int. Genet. 2017, 31, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Sturk, K.A.; Coble, M.; Barritt, S.M.; Irwin, J.A. Evaluation of modified Yfiler amplification strategy for compromised samples. Croat. Med. J. 2009, 50, 228–238. [Google Scholar] [CrossRef]
- Gorden, E.M.; Greytak, E.M.; Sturk-Andreaggi, K.; Cady, J.; McMahon, T.P.; Armentrout, S.; Marshall, C. Extended kinship analysis of historical remains using SNP capture. Forensic Sci. Int. Genet. 2021, 57, 102636. [Google Scholar] [CrossRef]
- Grandell, I.; Samara, R.; Tillmar, A.O. A SNP panel for identity and kinship testing using massive parallel sequencing. Int. J. Leg. Med. 2016, 130, 905–914. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Parson, W.; Allen, M.; Marshall, C. Impact of the sequencing method on the detection and interpretation of mitochondrial DNA length heteroplasmy. Forensic Sci. Int. Genet. 2019, 44, 102205. [Google Scholar] [CrossRef]
- Knapp, M.; Hofreiter, M. Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives. Genes 2010, 1, 227–243. [Google Scholar] [CrossRef]
- Edson, S.M. Extraction of DNA from Skeletonized Postcranial Remains: A Discussion of Protocols and Testing Modalities. J. Forensic Sci. 2019, 64, 1312–1323. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Context/Region | Number of Burials | Approximate Years Postmortem |
|---|---|---|
| JB55, Griswold, Connecticut, USA | 2 | 200 |
| Arch Street, Philadelphia, Pennsylvania, USA | 9 | 275 |
| World War II-Germany | 3 | 80 |
| World War II-Punchbowl 1, Honolulu, Hawaii, USA | 2 | 80 |
| Harewood Cemetery, Charles Town, West Virginia, USA | 3 | 200 |
| Basilica of St. Mary’s Assumption, Fürstenfeldbruck, Germany | 12 | 700–800 |
| Kit | Small Autosomal Target Size | Large Autosomal Target Size | Y-Chromosomal Target Size(s) | IPC Target Size |
|---|---|---|---|---|
| PowerQuant System | 84 bp | 294 bp | 81 bp, 136 bp | 435 bp |
| Investigator Quantiplex Pro | 91 bp | 353 bp | 81 bp | 434 bp |
| Quantifiler Trio | 80 bp | 214 bp | 75 bp | 130 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.T.; Cavagnino, C.; Kjelland, K.; Anderson, E.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Amory, C.; Spatola, B.; Moran, K.; Parson, W.; et al. Evaluating the Usefulness of Human DNA Quantification to Predict DNA Profiling Success of Historical Bone Samples. Genes 2023, 14, 994. https://doi.org/10.3390/genes14050994
Thomas JT, Cavagnino C, Kjelland K, Anderson E, Sturk-Andreaggi K, Daniels-Higginbotham J, Amory C, Spatola B, Moran K, Parson W, et al. Evaluating the Usefulness of Human DNA Quantification to Predict DNA Profiling Success of Historical Bone Samples. Genes. 2023; 14(5):994. https://doi.org/10.3390/genes14050994
Chicago/Turabian StyleThomas, Jacqueline Tyler, Courtney Cavagnino, Katelyn Kjelland, Elise Anderson, Kimberly Sturk-Andreaggi, Jennifer Daniels-Higginbotham, Christina Amory, Brian Spatola, Kimberlee Moran, Walther Parson, and et al. 2023. "Evaluating the Usefulness of Human DNA Quantification to Predict DNA Profiling Success of Historical Bone Samples" Genes 14, no. 5: 994. https://doi.org/10.3390/genes14050994
APA StyleThomas, J. T., Cavagnino, C., Kjelland, K., Anderson, E., Sturk-Andreaggi, K., Daniels-Higginbotham, J., Amory, C., Spatola, B., Moran, K., Parson, W., & Marshall, C. (2023). Evaluating the Usefulness of Human DNA Quantification to Predict DNA Profiling Success of Historical Bone Samples. Genes, 14(5), 994. https://doi.org/10.3390/genes14050994