
Redox-Based Strategies against Infections by Eukaryotic Pathogens

 ,
,
Abstract
1. Introduction, Scope, and Aims of This Review
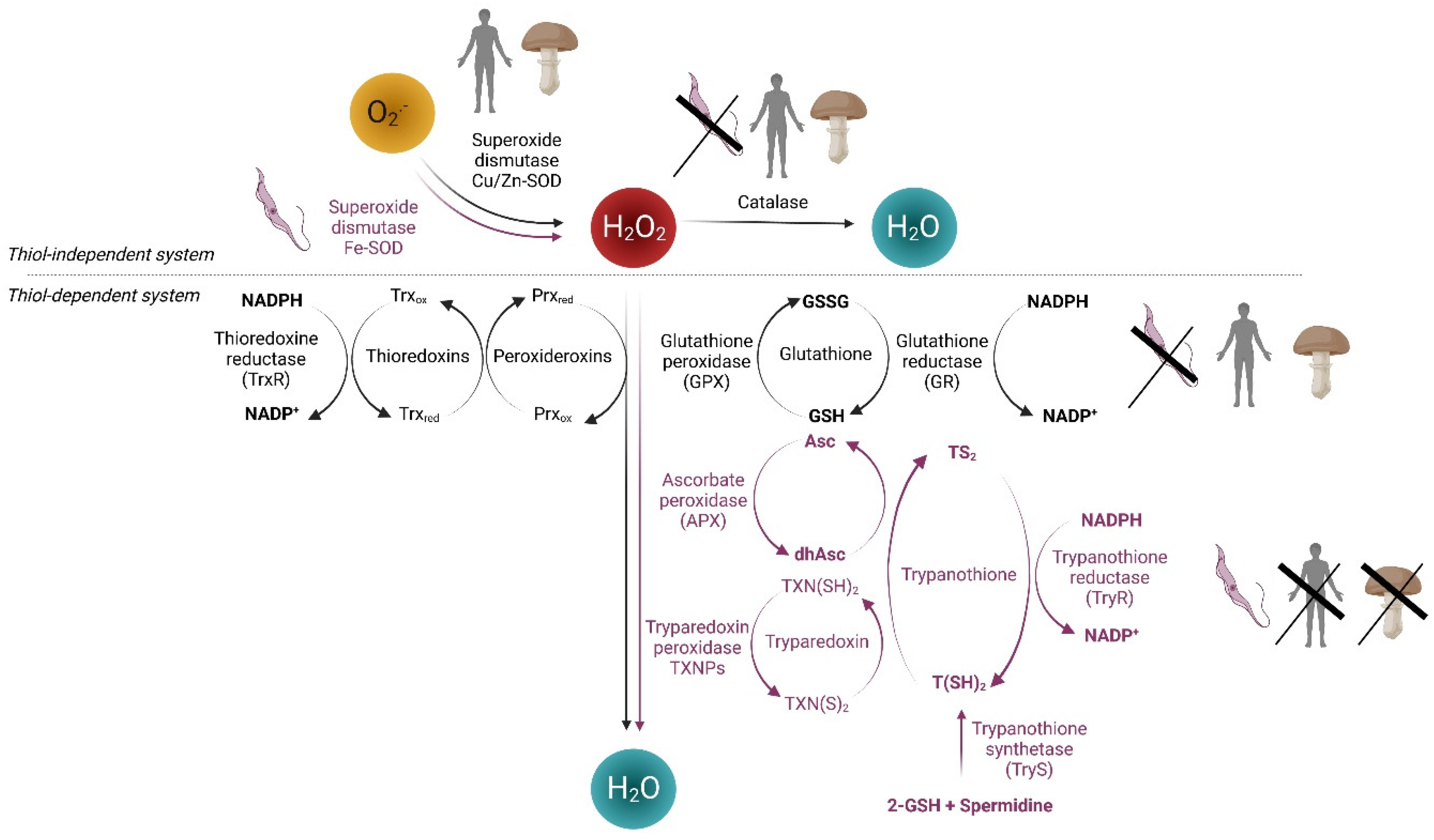
1.1. Reactive Oxygen Species (ROS) and Redox System in Human Cells
1.2. Redox Metabolism of Pathogenic Fungi
1.3. Redox Metabolism in Protozoan Parasites
2. Targeting Redox Specificities in Parasites
2.1. Targeting Fe-SOD
2.2. Targeting Trypanothione
3. Targeting the Thioredoxin Reductase TrxR
4. Targeting the Fungal TCA Cycle to Perturb the Redox Balance
5. Targeting Fungi through ROS Production

{kind=link}
| Name of the Molecule | Target | Mode of Action | References |
|---|---|---|---|
| Amphotericin B | Fungal membrane | Ergosterol binding | [75] |
| Artemisinin | NADH dehydrogenases | Oxidative stress generation | [63,65,66] |
| Auranofin | Thioredoxin reductases; Mia40/Erv1 import relay | Anti-oxidative stress response pathway | [37,38,39,40] |
| Bithionol | Adenylyl cyclase | cAMP synthesis inhibition | [76] |
| 5-methyl-phenazine-1-carboxylic acid (5MPCA) | ROS production | [74] | |
| 5-nitroimidazoles | Cysteines in TrxR | TrxR activity inhibition | [30,31] |
| Isoxanthohumol | TCA cycle and respiration | ATP production decreased, ROS production increased | [72] |
| Itraconazole | Fungal membrane | Ergosterol binding | [75] |
| Mannich base-type compounds | Fe-SOD | Fe-SOD inhibition | [19,20] |
| Melarsoprol | Pyruvate kinase enzyme | ||
| Metallic nanoparticles | ROS production | [78,79,80,81] | |
| Methylaervine | TCA cycle, steroid biosynthesis | Redox balance disruptuin | [73] |
| N-(6-quinolinemethyl)-3-pyrazole carboxamide | LmTR | LmTR inhibition | [23,26] |
| Plasmodione | NADH dehydrogenases | Oxidative stress generation | [68,69] |
| Tetradentate polyamines | Fe-SOD | Fe-SOD inhibition | [18] |
| Triazole analogues | LiTR | Slow-binding inhibitors of LiTryR | [27] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug. Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Sun, S.; Hoy, M.J.; Heitman, J. Fungal pathogens. Curr. Biol. 2020, 30, R1163–R1169. [Google Scholar] [CrossRef]
- Brown, A.J.; Haynes, K.; Quinn, J. Nitrosative and oxidative stress responses in fungal pathogenicity. Curr. Opin. Microbiol. 2009, 12, 384–391. [Google Scholar] [CrossRef]
- Yaakoub, H.; Mina, S.; Calenda, A.; Bouchara, J.P.; Papon, N. Oxidative stress response pathways in fungi. Cell. Mol. Life Sci. 2022, 79, 333. [Google Scholar] [CrossRef]
- Ali, V.; Behera, S.; Nawaz, A.; Equbal, A.; Pandey, K. Unique thiol metabolism in trypanosomatids: Redox homeostasis and drug resistance. Adv. Parasitol. 2022, 117, 75–155. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Blackburn, P.; Ulrich, P.; Chait, B.T.; Cerami, A. Trypanothione: A Novel Bis(glutathionyl)spermidine Cofactor for Glutathione Reductase in Trypanosomatids. Science 1985, 227, 1485–1487. [Google Scholar] [CrossRef]
- Santi, A.M.M.; Murta, S.M.F. Antioxidant defence system as a rational target for Chagas disease and Leishmaniasis chemotherapy. Mem. Do Inst. Oswaldo Cruz 2022, 117, e210401. [Google Scholar] [CrossRef] [PubMed]
- Egwu, C.O.; Augereau, J.M.; Reybier, K.; Benoit-Vical, F. Reactive Oxygen Species as the Brainbox in Malaria Treatment. Antioxidants 2021, 10, 1872. [Google Scholar] [CrossRef] [PubMed]
- Fichera, M.E.; Roos, D.S. A plastid organelle as a drug target in apicomplexan parasites. Nature 1997, 390, 407–409. [Google Scholar] [CrossRef]
- Jeelani, G.; Nozaki, T. Entamoeba thiol-based redox metabolism: A potential target for drug development. Mol. Biochem. Parasitol. 2016, 206, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Radheshyam, M.; Madhulika, N. Superoxide Dismutase: A Key Enzyme for the Survival of Intracellular Pathogens in Host. In Reactive Oxygen Species; Rizwan, A., Ed.; IntechOpen: Rijeka, 2021; p. Ch. 3. [Google Scholar]
- Sánchez-Moreno, M.; Gómez-Contreras, F.; Navarro, P.; Marín, C.; Olmo, F.; Yunta, M.J.; Sanz, A.M.; Rosales, M.J.; Cano, C.; Campayo, L. Phthalazine derivatives containing imidazole rings behave as Fe-SOD inhibitors and show remarkable anti-T. cruzi activity in immunodeficient-mouse mode of infection. J. Med. Chem. 2012, 55, 9900–9913. [Google Scholar] [CrossRef]
- Santi, A.M.M.; Silva, P.A.; Santos, I.F.M.; Murta, S.M.F. Downregulation of FeSOD-A expression in Leishmania infantum alters trivalent antimony and miltefosine susceptibility. Parasit. Vectors 2021, 14, 366. [Google Scholar] [CrossRef]
- Urbanová, K.; Ramírez-Macías, I.; Martín-Escolano, R.; Rosales, M.J.; Cussó, O.; Serrano, J.; Company, A.; Sánchez-Moreno, M.; Costas, M.; Ribas, X.; et al. Effective Tetradentate Compound Complexes against Leishmania spp. that Act on Critical Enzymatic Pathways of These Parasites. Molecules 2018, 24, 134. [Google Scholar] [CrossRef]
- Martín-Escolano, R.; Moreno-Viguri, E.; Santivañez-Veliz, M.; Martin-Montes, A.; Medina-Carmona, E.; Paucar, R.; Marín, C.; Azqueta, A.; Cirauqui, N.; Pey, A.L.; et al. Second Generation of Mannich Base-Type Derivatives with in Vivo Activity against Trypanosoma cruzi. J. Med. Chem. 2018, 61, 5643–5663. [Google Scholar] [CrossRef]
- Paucar, R.; Martín-Escolano, R.; Moreno-Viguri, E.; Azqueta, A.; Cirauqui, N.; Marín, C.; Sánchez-Moreno, M.; Pérez-Silanes, S. Rational modification of Mannich base-type derivatives as novel antichagasic compounds: Synthesis, in vitro and in vivo evaluation. Bioorg. Med. Chem. 2019, 27, 3902–3917. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Horn, D. Melarsoprol Resistance in African Trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef]
- Kumar, A.; Chauhan, N.; Singh, S. Understanding the Cross-Talk of Redox Metabolism and Fe-S Cluster Biogenesis in Leishmania Through Systems Biology Approach. Front. Cell. Infect. Microbiol. 2019, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Matadamas-Martínez, F.; Hernández-Campos, A.; Téllez-Valencia, A.; Vázquez-Raygoza, A.; Comparán-Alarcón, S.; Yépez-Mulia, L.; Castillo, R. Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules 2019, 24, 3216. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D. Amphotericin B: Spectrum and resistance. J. Antimicrob. Chemother. 2002, 49, 7–10. [Google Scholar] [CrossRef] [PubMed]
- den Boer, M.; Davidson, R.N. Treatment options for visceral leishmaniasis. Expert. Rev. Anti-Infect. Ther. 2006, 4, 187–197. [Google Scholar] [CrossRef]
- Espinosa-Bustos, C.; Ortiz Pérez, M.; Gonzalez-Gonzalez, A.; Zarate, A.M.; Rivera, G.; Belmont-Díaz, J.A.; Saavedra, E.; Cuellar, M.A.; Vázquez, K.; Salas, C.O. New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase. Pharmaceutics 2022, 14, 1121. [Google Scholar] [CrossRef]
- Revuelto, A.; de Lucio, H.; García-Soriano, J.C.; Sánchez-Murcia, P.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; Velázquez, S. Efficient Dimerization Disruption of Leishmania infantum Trypanothione Reductase by Triazole-phenyl-thiazoles. J. Med. Chem. 2021, 64, 6137–6160. [Google Scholar] [CrossRef]
- Fritz-Wolf, K.; Jortzik, E.; Stumpf, M.; Preuss, J.; Iozef, R.; Rahlfs, S.; Becker, K. Crystal structure of the Plasmodium falciparum thioredoxin reductase-thioredoxin complex. J. Mol. Biol. 2013, 425, 3446–3460. [Google Scholar] [CrossRef]
- Krnajski, Z.; Gilberger, T.W.; Walter, R.D.; Cowman, A.F.; Müller, S. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 2002, 277, 25970–25975. [Google Scholar] [CrossRef]
- Leitsch, D.; Kolarich, D.; Wilson, I.B.H.; Altmann, F.; Duchêne, M. Nitroimidazole Action in Entamoeba histolytica: A Central Role for Thioredoxin Reductase. PLoS Biol. 2007, 5, e211. [Google Scholar] [CrossRef]
- Leitsch, D.; Kolarich, D.; Binder, M.; Stadlmann, J.; Altmann, F.; Duchêne, M. Trichomonas vaginalis: Metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implications for nitroimidazole toxicity and resistance. Mol. Microbiol. 2009, 72, 518–536. [Google Scholar] [CrossRef]
- Leitsch, D.; Schlosser, S.; Burgess, A.; Duchêne, M. Nitroimidazole drugs vary in their mode of action in the human parasite Giardia lamblia. Int. J. Parasitol. Drugs Drug. Resist. 2012, 2, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.D.; Sadler, P.J.; Scawen, M.D.; Silver, S. Effects of gold(I) antiarthritic drugs and related compounds on Pseudomonas putida. J. Inorg. Biochem. 1992, 46, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Rosario, S.; Cowart, D.; Myers, A.; Tarrien, R.; Levine, R.L.; Scott, R.A.; Self, W.T. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2009, 14, 507–519. [Google Scholar] [CrossRef]
- Jackson-Rosario, S.; Self, W.T. Inhibition of selenium metabolism in the oral pathogen Treponema denticola. J. Bacteriol. 2009, 191, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P.; Patterson, T.F.; Srinivasan, A.; Chaturvedi, A.K.; Fothergill, A.W.; Wormley, F.L.; Ramasubramanian, A.K.; Lopez-Ribot, J.L. Repurposing auranofin as an antifungal: In vitro activity against a variety of medically important fungi. Virulence 2017, 8, 138–142. [Google Scholar] [CrossRef]
- Fuchs, B.B.; RajaMuthiah, R.; Souza, A.C.; Eatemadpour, S.; Rossoni, R.D.; Santos, D.A.; Junqueira, J.C.; Rice, L.B.; Mylonakis, E. Inhibition of bacterial and fungal pathogens by the orphaned drug auranofin. Future Med. Chem. 2016, 8, 117–132. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs RD 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Thangamani, S.; Maland, M.; Mohammad, H.; Pascuzzi, P.E.; Avramova, L.; Koehler, C.M.; Hazbun, T.R.; Seleem, M.N. Repurposing Approach Identifies Auranofin with Broad Spectrum Antifungal Activity That Targets Mia40-Erv1 Pathway. Front. Cell. Infect. Microbiol. 2017, 7, 4. [Google Scholar] [CrossRef]
- Rossi, S.A.; de Oliveira, H.C.; Agreda-Mellon, D.; Lucio, J.; Mendes-Giannini, M.J.S.; García-Cambero, J.P.; Zaragoza, O. Identification of Off-Patent Drugs That Show Synergism with Amphotericin B or That Present Antifungal Action against Cryptococcus neoformans and Candida spp. Antimicrob. Agents Chemother. 2020, 64, e01921-19. [Google Scholar] [CrossRef]
- Capparelli, E.V.; Bricker-Ford, R.; Rogers, M.J.; McKerrow, J.H.; Reed, S.L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob. Agents Chemother. 2017, 61, e01947-16. [Google Scholar] [CrossRef]
- Debnath, A.; Parsonage, D.; Andrade, R.M.; He, C.; Cobo, E.R.; Hirata, K.; Chen, S.; Garcia-Rivera, G.; Orozco, E.; Martinez, M.B.; et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat. Med. 2012, 18, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Harbut, M.B.; Vilcheze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R., Jr.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, D.; Sheng, F.; Hirata, K.; Debnath, A.; McKerrow, J.H.; Reed, S.L.; Abagyan, R.; Poole, L.B.; Podust, L.M. X-ray structures of thioredoxin and thioredoxin reductase from Entamoeba histolytica and prevailing hypothesis of the mechanism of Auranofin action. J. Struct. Biol. 2016, 194, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Thangamani, S.; Mohammad, H.; Abushahba, M.F.; Sobreira, T.J.; Hedrick, V.E.; Paul, L.N.; Seleem, M.N. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci. Rep. 2016, 6, 22571. [Google Scholar] [CrossRef] [PubMed]
- Sannella, A.R.; Casini, A.; Gabbiani, C.; Messori, L.; Bilia, A.R.; Vincieri, F.F.; Majori, G.; Severini, C. New uses for old drugs. Auranofin, a clinically established antiarthritic metallodrug, exhibits potent antimalarial effects in vitro: Mechanistic and pharmacological implications. FEBS Lett. 2008, 582, 844–847. [Google Scholar] [CrossRef]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; Di Muccio, T.; Colotti, G. A gold-containing drug against parasitic polyamine metabolism: The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids 2012, 42, 803–811. [Google Scholar] [CrossRef]
- Angelucci, F.; Sayed, A.A.; Williams, D.L.; Boumis, G.; Brunori, M.; Dimastrogiovanni, D.; Miele, A.E.; Pauly, F.; Bellelli, A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: Structural and kinetic aspects. J. Biol. Chem. 2009, 284, 28977–28985. [Google Scholar] [CrossRef]
- Abhishek, S.; Sivadas, S.; Satish, M.; Deeksha, W.; Rajakumara, E. Dynamic Basis for Auranofin Drug Recognition by Thiol-Reductases of Human Pathogens and Intermediate Coordinated Adduct Formation with Catalytic Cysteine Residues. ACS Omega 2019, 4, 9593–9602. [Google Scholar] [CrossRef]
- Leitsch, D.; Müller, J.; Müller, N. Evaluation of Giardia lamblia thioredoxin reductase as drug activating enzyme and as drug target. Int. J. Parasitol. Drugs Drug. Resist. 2016, 6, 148–153. [Google Scholar] [CrossRef]
- Leitsch, D. Drug susceptibility testing in microaerophilic parasites: Cysteine strongly affects the effectivities of metronidazole and auranofin, a novel and promising antimicrobial. Int. J. Parasitol. Drugs Drug. Resist. 2017, 7, 321–327. [Google Scholar] [CrossRef]
- Ma, C.I.; Tirtorahardjo, J.A.; Jan, S.; Schweizer, S.S.; Rosario, S.A.C.; Du, Y.; Zhang, J.J.; Morrissette, N.S.; Andrade, R.M. Auranofin Resistance in Toxoplasma gondii Decreases the Accumulation of Reactive Oxygen Species but Does Not Target Parasite Thioredoxin Reductase. Front. Cell. Infect. Microbiol. 2021, 11, 618994. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Su, X. Artemisinin: Discovery from the Chinese herbal garden. Cell 2011, 146, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Wittlin, S.; Mäser, P. From Magic Bullet to Magic Bomb: Reductive Bioactivation of Antiparasitic Agents. ACS Infect. Dis. 2021, 7, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Wittlin, S.; Nehrbass-Stuedli, A.; Dong, Y.; Wang, X.; Hemphill, A.; Matile, H.; Brun, R.; Vennerstrom, J.L. Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160). Antimicrob. Agents Chemother. 2007, 51, 2991–2993. [Google Scholar] [CrossRef] [PubMed]
- Charman, S.A.; Arbe-Barnes, S.; Bathurst, I.C.; Brun, R.; Campbell, M.; Charman, W.N.; Chiu, F.C.; Chollet, J.; Craft, J.C.; Creek, D.J.; et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci. USA 2011, 108, 4400–4405. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Hammill, J.T.; Guy, R.K. Seeking the Elusive Long-Acting Ozonide: Discovery of Artefenomel (OZ439). J. Med. Chem. 2017, 60, 2651–2653. [Google Scholar] [CrossRef]
- Siddiqui, G.; Giannangelo, C.; De Paoli, A.; Schuh, A.K.; Heimsch, K.C.; Anderson, D.; Brown, T.G.; MacRaild, C.A.; Wu, J.; Wang, X.; et al. Peroxide Antimalarial Drugs Target Redox Homeostasis in Plasmodium falciparum Infected Red Blood Cells. ACS Infect. Dis. 2022, 8, 210–226. [Google Scholar] [CrossRef]
- Egwu, C.O.; Pério, P.; Augereau, J.M.; Tsamesidis, I.; Benoit-Vical, F.; Reybier, K. Resistance to artemisinin in falciparum malaria parasites: A redox-mediated phenomenon. Free Radic. Biol. Med. 2022, 179, 317–327. [Google Scholar] [CrossRef]
- Tan, R.X.; Lu, H.; Wolfender, J.L.; Yu, T.T.; Zheng, W.F.; Yang, L.; Gafner, S.; Hostettmann, K. Mono- and sesquiterpenes and antifungal constituents from Artemisia species. Planta Med. 1999, 65, 64–67. [Google Scholar] [CrossRef]
- Galal, A.M.; Ross, S.A.; Jacob, M.; ElSohly, M.A. Antifungal activity of artemisinin derivatives. J. Nat. Prod. 2005, 68, 1274–1276. [Google Scholar] [CrossRef]
- Dhingra, V.; Pakki, S.R.; Narasu, M.L. Antimicrobial activity of artemisinin and its precursors. Curr. Sci. 2000, 78, 709–713. [Google Scholar]
- Li, W.; Mo, W.; Shen, D.; Sun, L.; Wang, J.; Lu, S.; Gitschier, J.M.; Zhou, B. Yeast model uncovers dual roles of mitochondria in action of artemisinin. PLoS Genet. 2005, 1, e36. [Google Scholar] [CrossRef] [PubMed]
- Gautam, P.; Upadhyay, S.K.; Hassan, W.; Madan, T.; Sirdeshmukh, R.; Sundaram, C.S.; Gade, W.N.; Basir, S.F.; Singh, Y.; Sarma, P.U. Transcriptomic and proteomic profile of Aspergillus fumigatus on exposure to artemisinin. Mycopathologia 2011, 172, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, L.; Li, J.; Fan, Q.; Long, Y.; Li, Y.; Zhou, B. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS ONE 2010, 5, e9582. [Google Scholar] [CrossRef]
- Kimata-Ariga, Y.; Morihisa, R. Effect of Artemisinin on the Redox System of NADPH/FNR/Ferredoxin from Malaria Parasites. Antioxidants 2022, 11, 273. [Google Scholar] [CrossRef]
- Cichocki, B.A.; Donzel, M.; Heimsch, K.C.; Lesanavičius, M.; Feng, L.; Montagut, E.J.; Becker, K.; Aliverti, A.; Elhabiri, M.; Čėnas, N.; et al. Plasmodium falciparum Ferredoxin-NADP(+) Reductase-Catalyzed Redox Cycling of Plasmodione Generates Both Predicted Key Drug Metabolites: Implication for Antimalarial Drug Development. ACS Infect. Dis. 2021, 7, 1996–2012. [Google Scholar] [CrossRef]
- Mounkoro, P.; Michel, T.; Golinelli-Cohen, M.P.; Blandin, S.; Davioud-Charvet, E.; Meunier, B. A role for the succinate dehydrogenase in the mode of action of the redox-active antimalarial drug, plasmodione. Free Radic. Biol. Med. 2021, 162, 533–541. [Google Scholar] [CrossRef]
- Mounkoro, P.; Michel, T.; Blandin, S.; Golinelli-Cohen, M.P.; Davioud-Charvet, E.; Meunier, B. Investigating the mode of action of the redox-active antimalarial drug plasmodione using the yeast model. Free Radic. Biol. Med. 2019, 141, 269–278. [Google Scholar] [CrossRef]
- Ke, H.; Lewis, I.A.; Morrisey, J.M.; McLean, K.J.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Jacobs-Lorena, M.; Llinas, M.; Vaidya, A.B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell. Rep. 2015, 11, 164–174. [Google Scholar] [CrossRef]
- Laleve, A.; Vallieres, C.; Golinelli-Cohen, M.P.; Bouton, C.; Song, Z.; Pawlik, G.; Tindall, S.M.; Avery, S.V.; Clain, J.; Meunier, B. The antimalarial drug primaquine targets Fe-S cluster proteins and yeast respiratory growth. Redox Biol. 2016, 7, 21–29. [Google Scholar] [CrossRef]
- Yan, Y.F.; Wu, T.L.; Du, S.S.; Wu, Z.R.; Hu, Y.M.; Zhang, Z.J.; Zhao, W.B.; Yang, C.J.; Liu, Y.Q. The Antifungal Mechanism of Isoxanthohumol from Humulus lupulus Linn. Int. J. Mol. Sci. 2021, 22, 853. [Google Scholar] [CrossRef] [PubMed]
- Dan, W.; Gao, J.; Li, L.; Xu, Y.; Wang, J.; Dai, J. Cellular and non-target metabolomics approaches to understand the antifungal activity of methylaervine against Fusarium solani. Bioorg. Med. Chem. Lett. 2021, 43, 128068. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.K.; Jacobs, N.J.; Rajamani, S.; Krishnamurthy, M.; Cubillos-Ruiz, J.R.; Hogan, D.A. Antifungal mechanisms by which a novel Pseudomonas aeruginosa phenazine toxin kills Candida albicans in biofilms. Mol. Microbiol. 2010, 78, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Chan, K.L.; Faria, N.C.; Martins Mde, L.; Campbell, B.C. Targeting the oxidative stress response system of fungi with redox-potent chemosensitizing agents. Front. Microbiol. 2012, 3, 88. [Google Scholar] [CrossRef]
- Kim, J.H.; Chan, K.L.; Cheng, L.W.; Tell, L.A.; Byrne, B.A.; Clothier, K.; Land, K.M. High Efficiency Drug Repurposing Design for New Antifungal Agents. Methods Protoc. 2019, 2, 31. [Google Scholar] [CrossRef]
- Pathakoti, K.; Manubolu, M.; Hwang, H.M. Nanostructures: Current uses and future applications in food science. J. Food Drug. Anal. 2017, 25, 245–253. [Google Scholar] [CrossRef]
- Slavin, Y.N.; Bach, H. Mechanisms of Antifungal Properties of Metal Nanoparticles. Nanomaterials 2022, 12, 4470. [Google Scholar] [CrossRef]
- Najibi Ilkhechi, N.; Mozammel, M.; Yari Khosroushahi, A. Antifungal effects of ZnO, TiO(2) and ZnO-TiO(2) nanostructures on Aspergillus flavus. Pestic. Biochem. Physiol. 2021, 176, 104869. [Google Scholar] [CrossRef]
- Abdal Dayem, A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef]
- Čapek, J.; Roušar, T. Detection of Oxidative Stress Induced by Nanomaterials in Cells-The Roles of Reactive Oxygen Species and Glutathione. Molecules 2021, 26, 4710. [Google Scholar] [CrossRef]
- Das, S.; Tripathy, S.; Pramanik, P.; Saha, B.; Roy, S. A novel nano-anti-malarial induces redox damage and elicits cytokine response to the parasite. Cytokine 2021, 144, 155555. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallières, C.; Golinelli-Cohen, M.-P.; Guittet, O.; Lepoivre, M.; Huang, M.-E.; Vernis, L. Redox-Based Strategies against Infections by Eukaryotic Pathogens. Genes 2023, 14, 778. https://doi.org/10.3390/genes14040778
Vallières C, Golinelli-Cohen M-P, Guittet O, Lepoivre M, Huang M-E, Vernis L. Redox-Based Strategies against Infections by Eukaryotic Pathogens. Genes. 2023; 14(4):778. https://doi.org/10.3390/genes14040778
Chicago/Turabian StyleVallières, Cindy, Marie-Pierre Golinelli-Cohen, Olivier Guittet, Michel Lepoivre, Meng-Er Huang, and Laurence Vernis. 2023. "Redox-Based Strategies against Infections by Eukaryotic Pathogens" Genes 14, no. 4: 778. https://doi.org/10.3390/genes14040778
APA StyleVallières, C., Golinelli-Cohen, M.-P., Guittet, O., Lepoivre, M., Huang, M.-E., & Vernis, L. (2023). Redox-Based Strategies against Infections by Eukaryotic Pathogens. Genes, 14(4), 778. https://doi.org/10.3390/genes14040778