
Nanopore-Based Direct RNA Sequencing of the Trypanosoma brucei Transcriptome Identifies Novel lncRNAs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Growth of Trypanosome Cells
2.2. Oligodeoxynucleotide Synthesis
2.3. RNA Isolation
2.4. Synthesis of cDNA and Quantitative Real-Time (qRT)-PCR
2.5. Preparation of DRS-Sequencing Libraries and Sequencing
2.6. Transcript Mapping
2.7. Identification of Full-Length Reads
2.8. Reference-Free Transcript Identification and Identification of lncRNA
2.9. Estimation of Transcript Abundance, DGE Analysis, and Statistics
3. Results and Discussion
3.1. RNA Sequencing Libraries—Quality Assessment
3.2. Identification of Full-Length Transcripts
3.3. Reference-Free Transcript Identification
3.4. Comparison with Annotated Exons
3.5. Long Noncoding RNAs
3.5.1. Characterisation of Long Noncoding RNAs
3.5.2. Novel lncRNAs
3.6. Differential Gene Expression (DGE) between Insect- and Bloodstream-Stage Trypanosomes
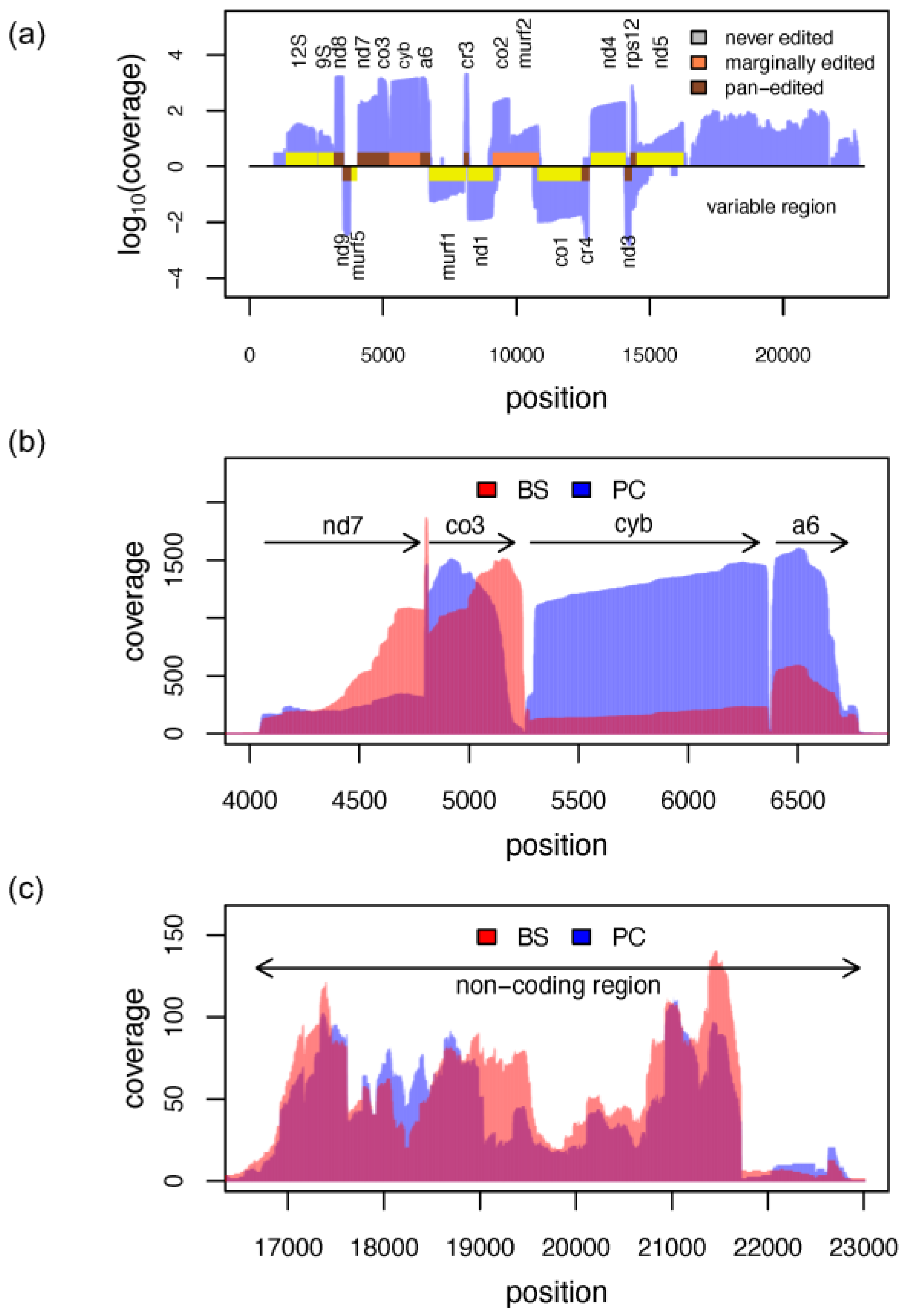
3.7. The Mitochondrial Transcriptome
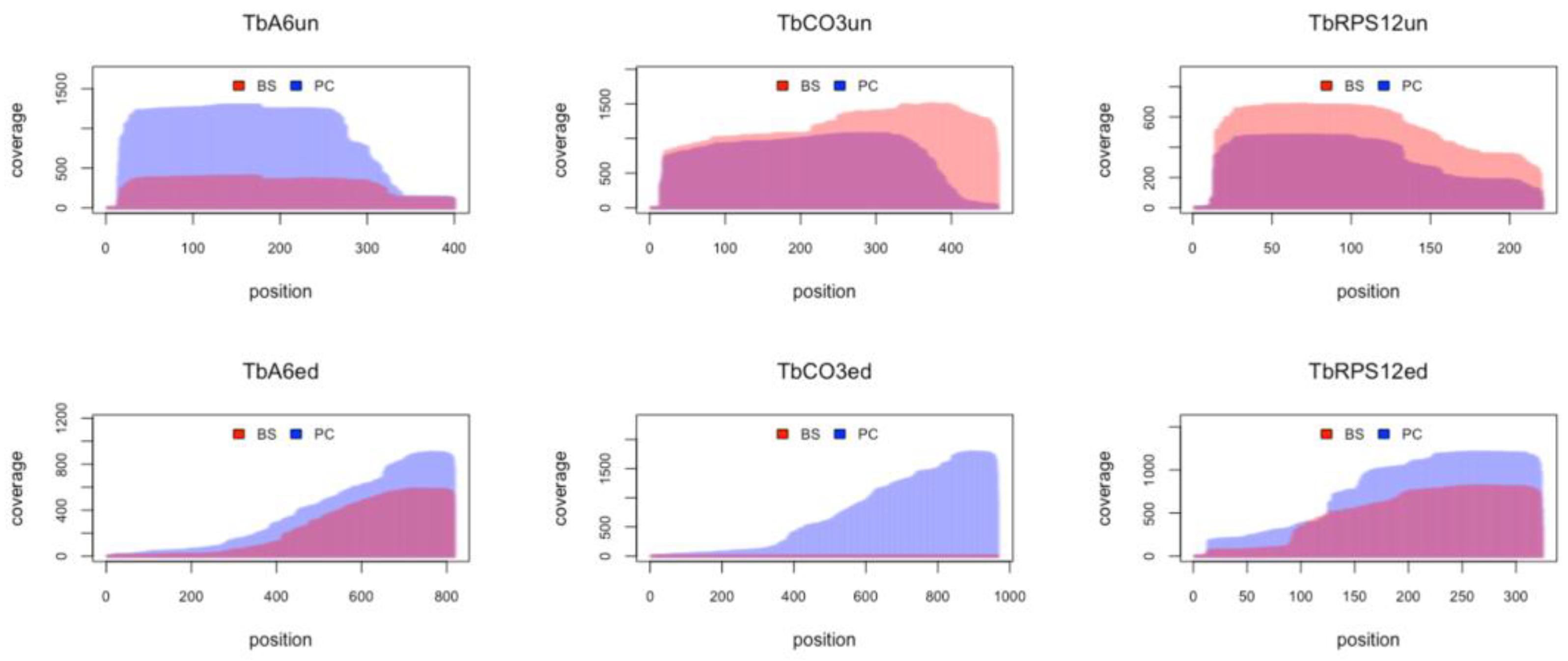
3.8. RNA Editing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaufer, A.; Stark, D.; Ellis, J. A review of the systematics, species identification and diagnostics of the Trypanosomatidae using the maxicircle kinetoplast DNA: From past to present. Int. J. Parasitol. 2020, 50, 449–460. [Google Scholar] [CrossRef] [PubMed]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: Progress and challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C. Regulation of gene expression in trypanosomatids: Living with polycistronic transcription. Open Biol. 2019, 9, 190072. [Google Scholar] [CrossRef] [PubMed]
- Kolev, N.G.; Ullu, E.; Tschudi, C. The emerging role of RNA-binding proteins in the life cycle of Trypanosoma brucei. Cell. Microbiol. 2014, 16, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Michaeli, S. Trans-splicing in trypanosomes: Machinery and its impact on the parasite transcriptome. Future Microbiol. 2011, 6, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Calvillo, S.; Florencio-Martínez, L.E.; Nepomuceno-Mejía, T. Nucleolar Structure and Function in Trypanosomatid Protozoa. Cells 2019, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.S.; Doniger, T.; Cohen-Chalamish, S.; Rengaraj, P.; Galili, B.; Aryal, S.; Unger, R.; Tschudi, C.; Michaeli, S. Developmentally Regulated Novel Non-coding Anti-sense Regulators of mRNA Translation in Trypanosoma brucei. iScience 2020, 23, 101780. [Google Scholar] [CrossRef]
- Kolev, N.G.; Rajan, K.S.; Tycowski, K.T.; Toh, J.Y.; Shi, H.; Lei, Y.; Michaeli, S.; Tschudi, C. The vault RNA of Trypanosoma brucei plays a role in the production of trans-spliced mRNA. J. Biol. Chem. 2019, 294, 15559–15574. [Google Scholar] [CrossRef]
- Michaeli, S.; Podell, D.; Agabian, N.; Ullu, E. The 7SL RNA homologue of Trypanosoma brucei is closely related to mammalian 7SL RNA. Mol. Biochem. Parasitol. 1992, 51, 55–64. [Google Scholar] [CrossRef]
- Sandhu, R.; Sanford, S.; Basu, S.; Park, M.; Pandya, U.M.; Li, B.; Chakrabarti, K. A trans-spliced telomerase RNA dictates telomere synthesis in Trypanosoma brucei. Cell Res. 2013, 23, 537–551. [Google Scholar] [CrossRef]
- Fort, R.S.; Chavez, S.; Trinidad Barnech, J.M.; Oliveira-Rizzo, C.; Smircich, P.; Sotelo-Silveira, J.R.; Duhagon, M.A. Current Status of Regulatory Non-Coding RNAs Research in the Tritryp. Noncoding RNA 2022, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Koslowsky, D.; Sun, Y.; Hindenach, J.; Theisen, T.; Lucas, J. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res. 2014, 42, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Kolev, N.G.; Franklin, J.B.; Carmi, S.; Shi, H.; Michaeli, S.; Tschudi, C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010, 6, e1001090. [Google Scholar] [CrossRef] [PubMed]
- Guegan, F.; Rajan, K.S.; Bento, F.; Pinto-Neves, D.; Sequeira, M.; Gumińska, N.; Mroczek, S.; Dziembowski, A.; Cohen-Chalamish, S.; Doniger, T.; et al. A long noncoding RNA promotes parasite differentiation in African trypanosomes. Sci. Adv. 2022, 8, eabn2706. [Google Scholar] [CrossRef]
- Minshall, N.; Git, A. Enzyme- and gene-specific biases in reverse transcription of RNA raise concerns for evaluating gene expression. Sci. Rep. 2020, 10, 8151. [Google Scholar] [CrossRef]
- Steijger, T.; Abril, J.F.; Engström, P.G.; Kokocinski, F.; RGASP Consortium; Hubbard, T.J.; Guigó, R.; Harrow, J.; Bertone, P. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef]
- Jan, C.H.; Friedman, R.C.; Ruby, J.G.; Bartel, D.P. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 2011, 469, 97–101. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS ONE 2010, 5, e12271. [Google Scholar] [CrossRef]
- Mourão, K.; Schurch, N.J.; Lucoszek, R.; Froussios, K.; MacKinnon, K.; Duc, C.; Simpson, G.; Barton, G.J. Detection and mitigation of spurious antisense expression with RoSA. F1000Research 2019, 8, 819. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Cross, G.A. Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei. Parasitology 1975, 71, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei bloodstream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Schönenberger, M. Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979, 36, 289–292. [Google Scholar]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Naguleswaran, A.; Fernandes, P.; Bevkal, S.; Rehmann, R.; Nicholson, P.; Roditi, I. Developmental changes and metabolic reprogramming during establishment of infection and progression of Trypanosoma brucei brucei through its insect host. PLoS Negl. Trop. Dis. 2021, 15, e0009504. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305, Erratum in: Nat. Methods 2020, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, Y.; Ma, Q.; Xu, Y.; Zhang, Y.; Du, W.; Wang, C.; Li, Y. LncFinder: An integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief Bioinform. 2019, 20, 2009–2027. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Jain, M.; Abu-Shumays, R.; Olsen, H.E.; Akeson, M. Advances in nanopore direct RNA sequencing. Nat. Methods 2022, 19, 1160–1164. [Google Scholar] [CrossRef]
- Krause, M.; Niazi, A.M.; Labun, K.; Torres Cleuren, Y.N.; Müller, F.S.; Valen, E. tailfindr: Alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA 2019, 25, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Maier, K.C.; Gressel, S.; Cramer, P.; Schwalb, B. Native molecule sequencing by nano-ID reveals synthesis and stability of RNA isoforms. Genome Res. 2020, 30, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Dawe, H.R.; Shaw, M.K.; Farr, H.; Gull, K. The hydrocephalus inducing gene product, Hydin, positions axonemal central pair microtubules. BMC Biol. 2007, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Fickett, J.W.; Tung, C.S. Assessment of protein coding measures. Nucleic Acids Res. 1992, 20, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Ersfeld, K.; Melville, S.E.; Gull, K. Nuclear and genome organization of Trypanosoma brucei. Parasitol. Today 1999, 15, 58–63. [Google Scholar] [CrossRef]
- Veitch, N.J.; Johnson, P.C.; Trivedi, U.; Terry, S.; Wildridge, D.; MacLeod, A. Digital gene expression analysis of two life cycle stages of the human-infective parasite, Trypanosoma brucei gambiense reveals differentially expressed clusters of co-regulated genes. BMC Genom. 2010, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Siegel, T.N.; Hekstra, D.R.; Wang, X.; Dewell, S.; Cross, G.A. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010, 38, 4946–4957. [Google Scholar] [CrossRef]
- Christiano, R.; Kolev, N.G.; Shi, H.; Ullu, E.; Walther, T.C.; Tschudi, C. The proteome and transcriptome of the infectious metacyclic form of Trypanosoma brucei define quiescent cells primed for mammalian invasion. Mol. Microbiol. 2017, 106, 74–92. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018, 46, e38. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.; Leger, A.; Prawer, Y.D.J.; Lane, T.A.; Harrison, P.J.; Haerty, W.; Clark, M.B. Accurate expression quantification from nanopore direct RNA sequencing with NanoCount. Nucleic Acids Res. 2022, 50, e19. [Google Scholar] [CrossRef]
- Bringaud, F.; Biteau, N.; Melville, S.E.; Hez, S.; El-Sayed, N.M.; Leech, V.; Berriman, M.; Hall, N.; Donelson, J.E.; Baltz, T. A new, expressed multigene family containing a hot spot for insertion of retroelements is associated with polymorphic subtelomeric regions of Trypanosoma brucei. Eukaryot. Cell 2002, 1, 137–151, Erratum in: Eukaryot. Cell 2002, 1, 305. [Google Scholar] [CrossRef]
- Bringaud, F.; Biteau, N.; Zuiderwijk, E.; Berriman, M.; El-Sayed, N.M.; Ghedin, E.; Melville, S.E.; Hall, N.; Baltz, T. The ingi and RIME non-LTR retrotransposons are not randomly distributed in the genome of Trypanosoma brucei. Mol. Biol. Evol. 2004, 21, 520–528. [Google Scholar] [CrossRef]
- Sement, F.M.; Suematsu, T.; Zhang, L.; Yu, T.; Huang, L.; Aphasizheva, I.; Aphasizhev, R. Transcription initiation defines kinetoplast RNA boundaries. Proc. Natl. Acad. Sci. USA 2018, 115, E10323–E10332. [Google Scholar] [CrossRef]
- Adler, B.K.; Harris, M.E.; Bertrand, K.I.; Hajduk, S.L. Modification of Trypanosoma brucei mitochondrial rRNA by posttranscriptional 3′ polyuridine tail formation. Mol. Cell. Biol. 1991, 11, 5878–5884. [Google Scholar]
- Aphasizhev, R.; Aphasizheva, I. Mitochondrial RNA processing in trypanosomes. Res. Microbiol. 2011, 162, 655–663. [Google Scholar] [CrossRef]
- Aphasizheva, I.; Alfonzo, J.; Carnes, J.; Cestari, I.; Cruz-Reyes, J.; Göringer, H.U.; Hajduk, S.; Lukeš, J.; Madison-Antenucci, S.; Maslov, D.A.; et al. Lexis and Grammar of Mitochondrial RNA Processing in Trypanosomes. Trends Parasitol. 2020, 36, 337–355. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Michels, P.A. Complex I of Trypanosomatidae: Does it exist? Trends Parasitol. 2008, 24, 310–317. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruse, E.; Göringer, H.U. Nanopore-Based Direct RNA Sequencing of the Trypanosoma brucei Transcriptome Identifies Novel lncRNAs. Genes 2023, 14, 610. https://doi.org/10.3390/genes14030610
Kruse E, Göringer HU. Nanopore-Based Direct RNA Sequencing of the Trypanosoma brucei Transcriptome Identifies Novel lncRNAs. Genes. 2023; 14(3):610. https://doi.org/10.3390/genes14030610
Chicago/Turabian StyleKruse, Elisabeth, and H. Ulrich Göringer. 2023. "Nanopore-Based Direct RNA Sequencing of the Trypanosoma brucei Transcriptome Identifies Novel lncRNAs" Genes 14, no. 3: 610. https://doi.org/10.3390/genes14030610
APA StyleKruse, E., & Göringer, H. U. (2023). Nanopore-Based Direct RNA Sequencing of the Trypanosoma brucei Transcriptome Identifies Novel lncRNAs. Genes, 14(3), 610. https://doi.org/10.3390/genes14030610