
SCP4ssd: A Serverless Platform for Nucleotide Sequence Synthesis Difficulty Prediction Using an AutoML Model
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets and Feature Extraction
2.2. Evaluation Metrics
2.3. Baseline Model Construction
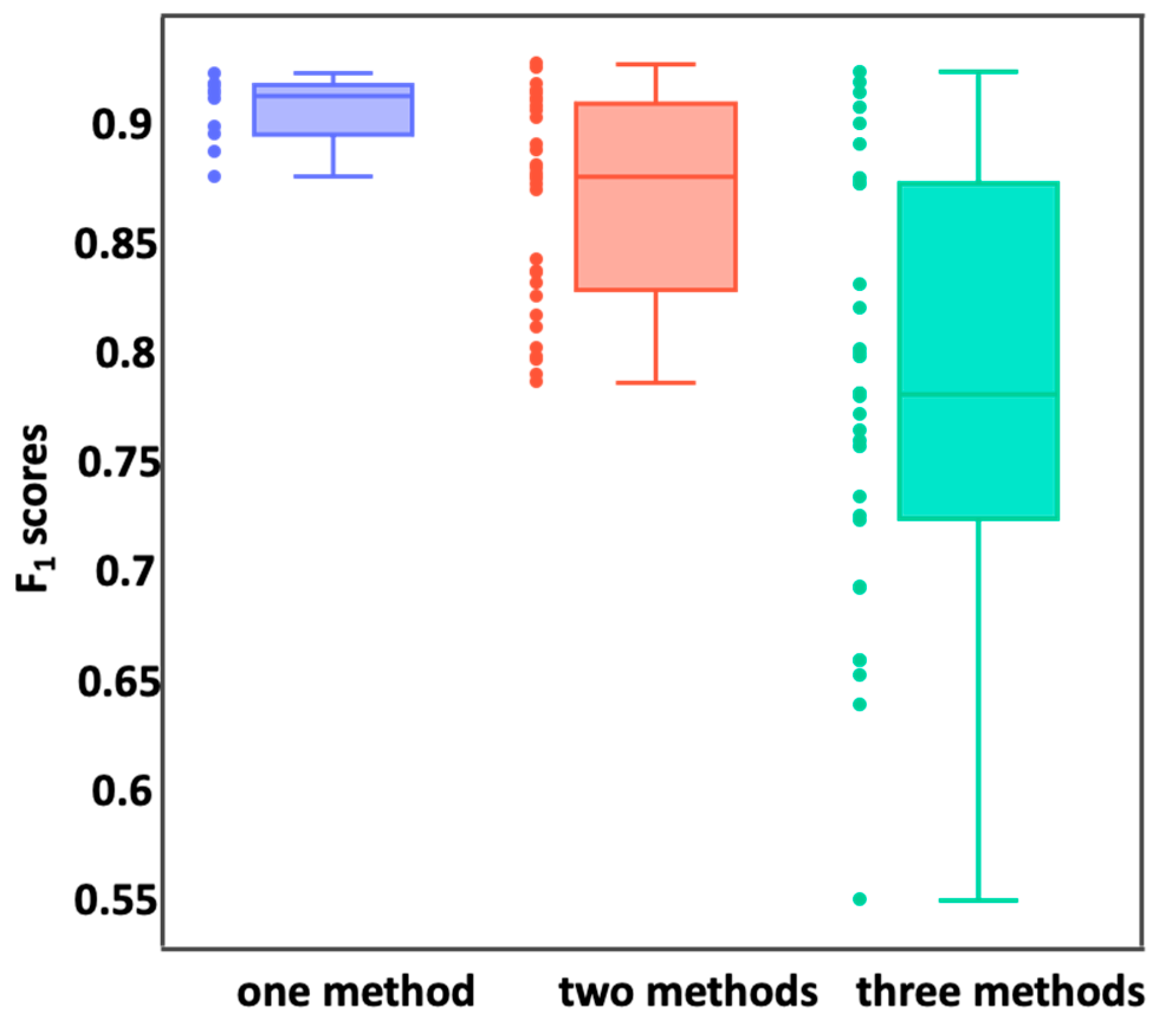
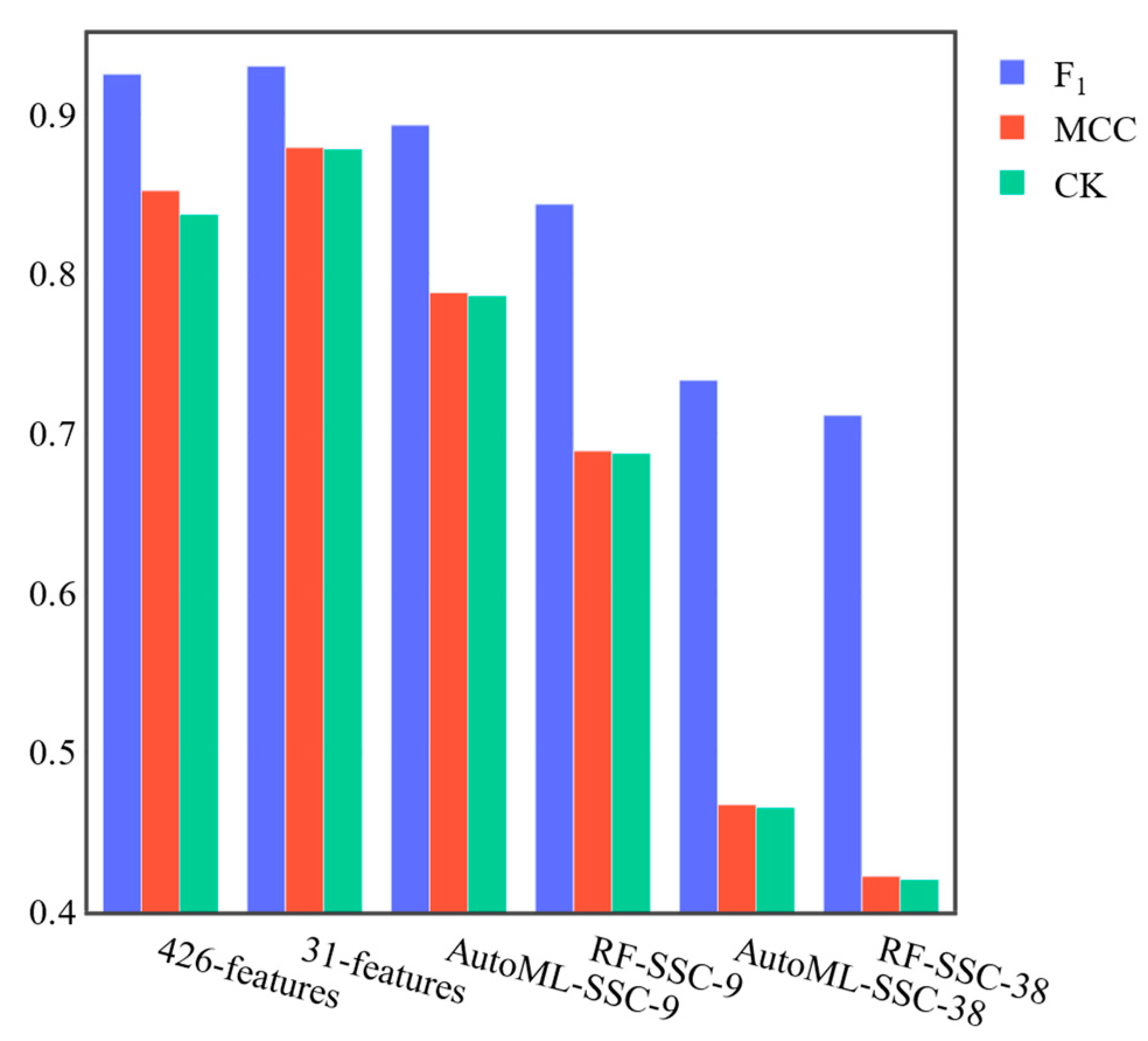
2.4. Feature Reduction
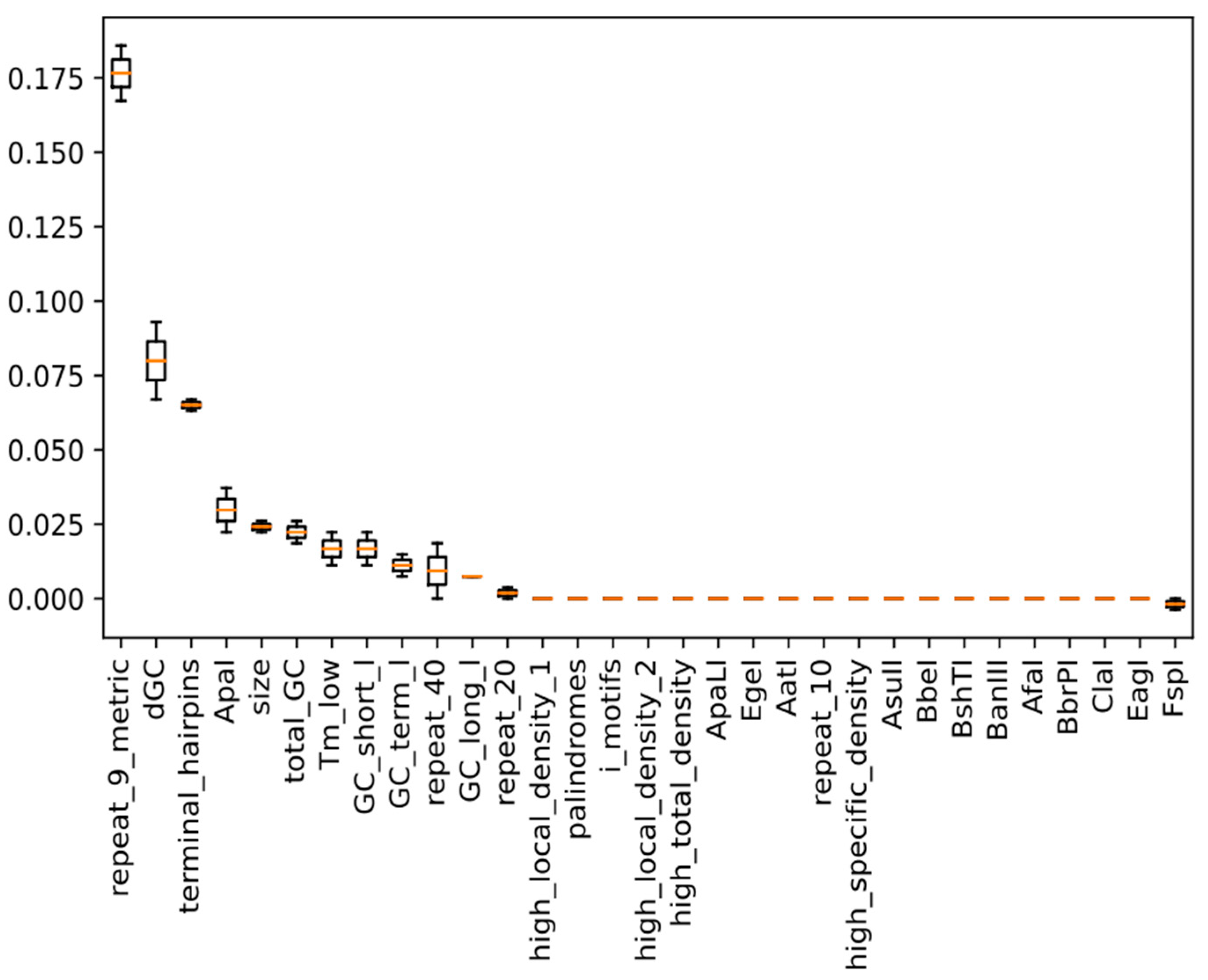
2.5. Model Training and Calculation of Feature Importance
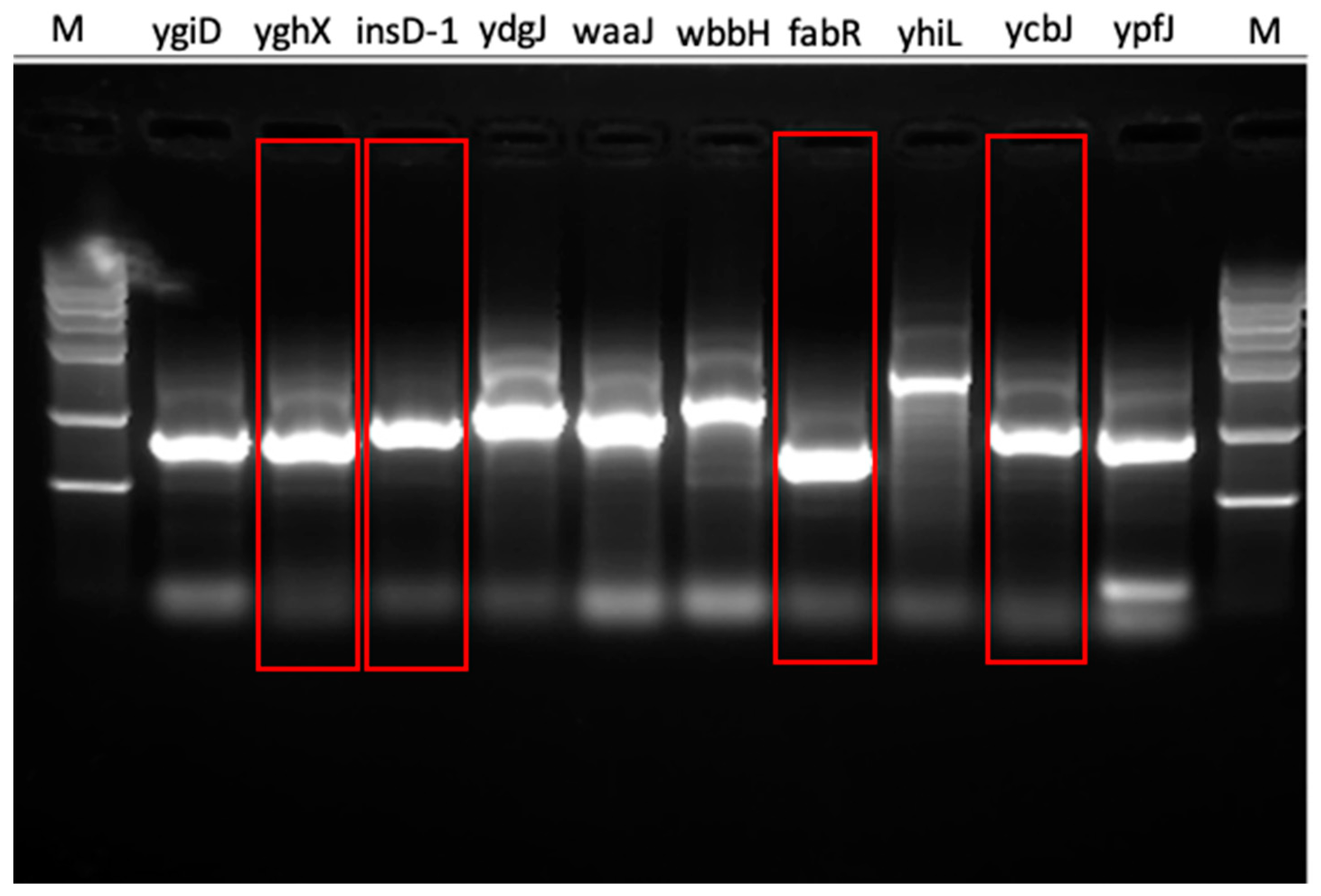
2.6. Experimental Validation
3. Results and Discussions
3.1. Baseline Model Construction
3.2. Feature Reduction
3.3. Benchmark Results of SCP4SSD and SSC
3.4. Experimental Validation
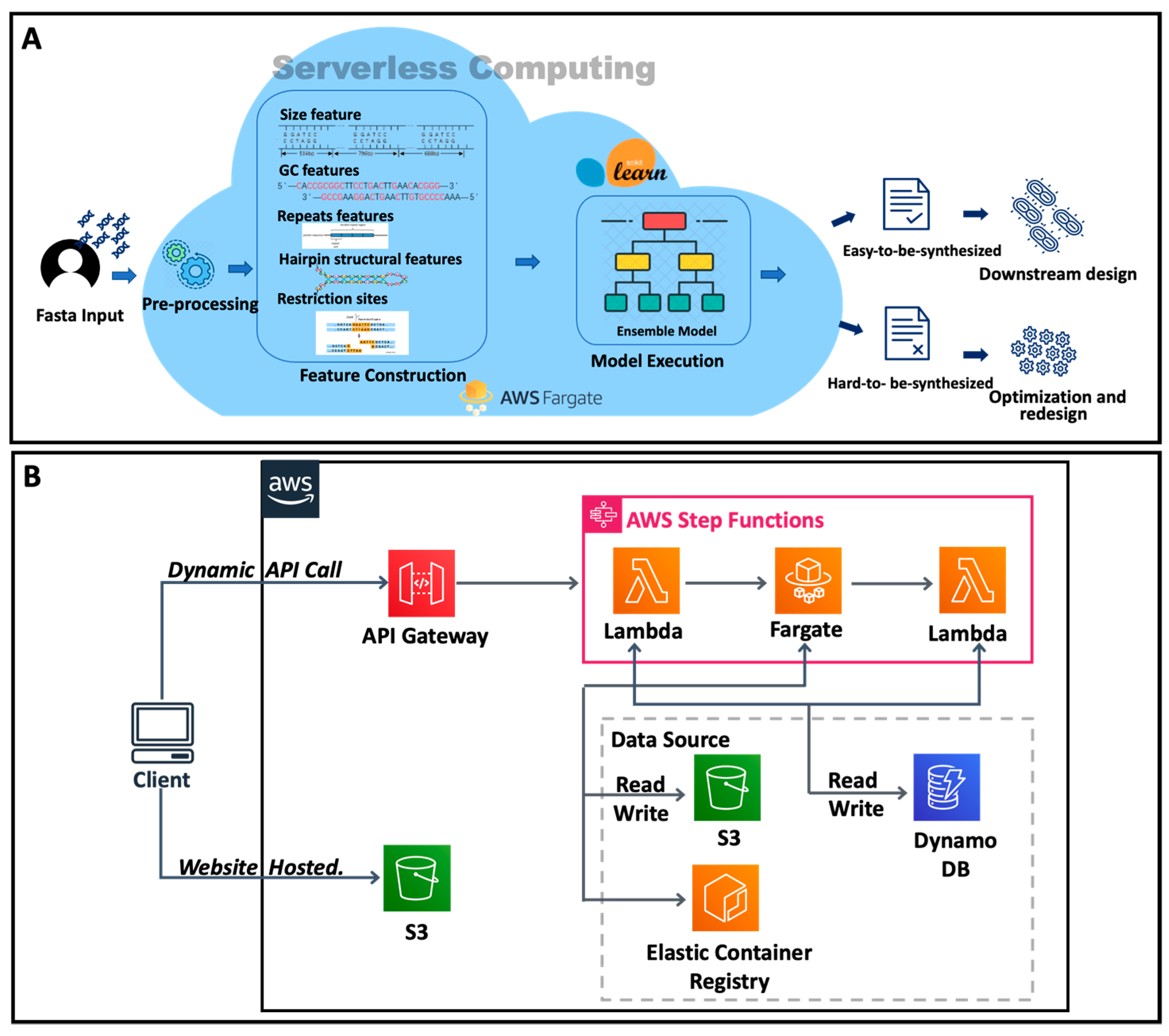
3.5. Cloud Platform
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, Z.; Yang, S.; Yuan, X.; Shi, Y.; Ouyang, L.; Jiang, S.; Yi, L.; Zhang, G. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis. Nucleic Acids Res. 2019, 47, e40. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, Z.; Wang, X.; Selvaraj, J.N.; Zhang, G. Genetic engineering modification and fermentation optimization for extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 2018, 102, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Li, X.; Zhang, R.; Yi, L.; Ma, Y.; Zhang, G. Tunnel engineering to accelerate product release for better biomass-degrading abilities in lignocellulolytic enzymes. Biotechnol. Biofuels 2019, 12, 275. [Google Scholar] [CrossRef]
- Park, J.W.; Lagniton, P.N.P.; Liu, Y.; Xu, R.H. mRNA vaccines for COVID-19: What, why and how. Int. J. Biol. Sci. 2021, 17, 1446–1460. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug. Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Annaluru, N.; Muller, H.; Mitchell, L.A.; Ramalingam, S.; Stracquadanio, G.; Richardson, S.M.; Dymond, J.S.; Kuang, Z.; Scheifele, L.Z.; Cooper, E.M.; et al. Total synthesis of a functional designer eukaryotic chromosome. Science 2014, 344, 55–58. [Google Scholar] [CrossRef]
- Hutchison, C.A., 3rd; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, aad6253. [Google Scholar] [CrossRef]
- Savina, K.; Sreekumar, R.; Soonu, V.K.; Variyar, E.J. Various vaccine platforms in the field of COVID-19. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 35. [Google Scholar] [CrossRef]
- Viswamitra, M.A.; Reddy, B.S.; Lin, G.H.; Sundaralingam, M. Stereochemistry of nucleic acids and their constituents. XVII. Crystal and molecular structure of deoxycytidine 5′-phosphate monohydrate. A possible puckering for the furanoside ring in B-deoxyribonucleic acid. J. Am. Chem. Soc. 1971, 93, 4565–4573. [Google Scholar] [CrossRef]
- Gibson, D.G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 2009, 37, 6984–6990. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.C.; Chilkoti, A. Combinatorial codon scrambling enables scalable gene synthesis and amplification of repetitive proteins. Nat. Mater. 2016, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G. Oligonucleotide assembly in yeast to produce synthetic DNA fragments. Methods Mol. Biol. 2012, 852, 11–21. [Google Scholar] [PubMed]
- Binkowski, B.F.; Richmond, K.E.; Kaysen, J.; Sussman, M.R.; Belshaw, P.J. Correcting errors in synthetic DNA through consensus shuffling. Nucleic Acids Res. 2005, 33, e55. [Google Scholar] [CrossRef]
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef]
- Roy, S.; Caruthers, M. Synthesis of DNA/RNA and their analogs via phosphoramidite and H-phosphonate chemistries. Molecules 2013, 18, 14268–14284. [Google Scholar] [CrossRef] [PubMed]
- Halper, S.M.; Hossain, A.; Salis, H.M. Synthesis Success Calculator: Predicting the Rapid Synthesis of DNA Fragments with Machine Learning. ACS Synth. Biol. 2020, 9, 1563–1571. [Google Scholar] [CrossRef]
- Ofer, D.; Linial, M. ProFET: Feature engineering captures high-level protein functions. Bioinformatics 2015, 31, 3429–3436. [Google Scholar] [CrossRef]
- Li, Z.R.; Lin, H.H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, W32–W37. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef]
- Bonidia, R.P.; Domingues, D.S.; Sanches, D.S.; de Carvalho, A. MathFeature: Feature extraction package for DNA, RNA and protein sequences based on mathematical descriptors. Brief. Bioinform. 2022, 23, bbab434. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Cang, Z.; Wei, G.W. A review of mathematical representations of biomolecular data. Phys. Chem. Chem. Phys. 2020, 22, 4343–4367. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.; Tamsir, A.; Voigt, C. Engineering bacterial signals and sensors. Contrib. Microbiol. 2009, 16, 194–225. [Google Scholar]
- Bonidia, R.P.; Sampaio, L.D.H.; Domingues, D.S.; Paschoal, A.R.; Lopes, F.M.; de Carvalho, A.; Sanches, D.S. Feature extraction approaches for biological sequences: A comparative study of mathematical features. Brief. Bioinform. 2021, 22, bbab011. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Liu, G.; Lu, D.; Lu, J. Pharm-AutoML: An open-source, end-to-end automated machine learning package for clinical outcome prediction. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 478–488. [Google Scholar] [CrossRef]
- Barreiro, E.; Munteanu, C.R.; Cruz-Monteagudo, M.; Pazos, A.; Gonzalez-Diaz, H. Net-Net Auto Machine Learning (AutoML) Prediction of Complex Ecosystems. Sci. Rep. 2018, 8, 12340. [Google Scholar] [CrossRef]
- Feurer, M.; Eggensperger, K.; Falkner, S.; Lindauer, M.; Hutter, F. Auto-sklearn 2.0: Hands-free automl via meta-learning. arXiv 2020, arXiv:200704074. [Google Scholar]
- Tang, J.; Alelyani, S.; Liu, H. Feature selection for classification: A review. In Data Classification: Algorithms and Applications; CRC Press: Boca Raton, FL, USA, 2014; pp. 37–64. [Google Scholar]
- Jović, A.; Brkić, K.; Bogunović, N. A review of feature selection methods with applications. In Proceedings of the 2015 38th International Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), Opatija, Croatia, 25–29 May 2015; pp. 1200–1205. [Google Scholar]
- Hoover, D.M.; Lubkowski, J. DNAWorks: An automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002, 30, e43. [Google Scholar] [CrossRef]
- Burkat, K.; Pawlik, M.; Balis, B.; Malawski, M.; Vahi, K.; Rynge, M.; Ferreira da Silva, R.; Deelman, E. Serverless Containers–rising viable approach to Scientific Workflows. In Proceedings of the 2021 IEEE 17th International Conference on eScience (eScience), Innsbruck, Austria, 20–23 September 2021; pp. 40–49. [Google Scholar]
- Carver, B.; Zhang, J.; Wang, A.; Anwar, A.; Wu, P.; Cheng, Y. Wukong: A scalable and locality-enhanced framework for serverless parallel computing. In Proceedings of the 11th ACM Symposium on Cloud Computing, Virtual Event, 19–21 October 2020; pp. 1–15. [Google Scholar]
- Jain, P.; Munjal, Y.; Gera, J.; Gupta, P. Performance analysis of various server hosting techniques. Procedia Comput. Sci. 2020, 173, 70–77. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | DNAWorks Score | Our Method | SSC | Experimental Result |
|---|---|---|---|---|
| ygiD | 6.485 | EASY | HARD | HARD |
| yghX | 6.756 | EASY | HARD | EASY |
| insD-1 | 4.512 | EASY | HARD | EASY |
| ydgJ | 6.5 | HARD | EASY | HARD |
| waaJ | 54.875 | HARD | EASY | HARD |
| wbbH | 64.955 | HARD | HARD | HARD |
| fabR | 6.084 | EASY | EASY | EASY |
| yhiL | 41.894 | HARD | EASY | HARD |
| ycbJ | 9.622 | EASY | EASY | EASY |
| ypfJ | 6.625 | EASY | HARD | HARD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ren, S.; Shi, Z.; Wang, R.; Li, H.; Tian, H.; Feng, M.; Liao, X.; Ma, H. SCP4ssd: A Serverless Platform for Nucleotide Sequence Synthesis Difficulty Prediction Using an AutoML Model. Genes 2023, 14, 605. https://doi.org/10.3390/genes14030605
Zhang J, Ren S, Shi Z, Wang R, Li H, Tian H, Feng M, Liao X, Ma H. SCP4ssd: A Serverless Platform for Nucleotide Sequence Synthesis Difficulty Prediction Using an AutoML Model. Genes. 2023; 14(3):605. https://doi.org/10.3390/genes14030605
Chicago/Turabian StyleZhang, Jianqi, Shuai Ren, Zhenkui Shi, Ruoyu Wang, Haoran Li, Huijuan Tian, Miao Feng, Xiaoping Liao, and Hongwu Ma. 2023. "SCP4ssd: A Serverless Platform for Nucleotide Sequence Synthesis Difficulty Prediction Using an AutoML Model" Genes 14, no. 3: 605. https://doi.org/10.3390/genes14030605
APA StyleZhang, J., Ren, S., Shi, Z., Wang, R., Li, H., Tian, H., Feng, M., Liao, X., & Ma, H. (2023). SCP4ssd: A Serverless Platform for Nucleotide Sequence Synthesis Difficulty Prediction Using an AutoML Model. Genes, 14(3), 605. https://doi.org/10.3390/genes14030605