
The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Pathogenesis of HGPS
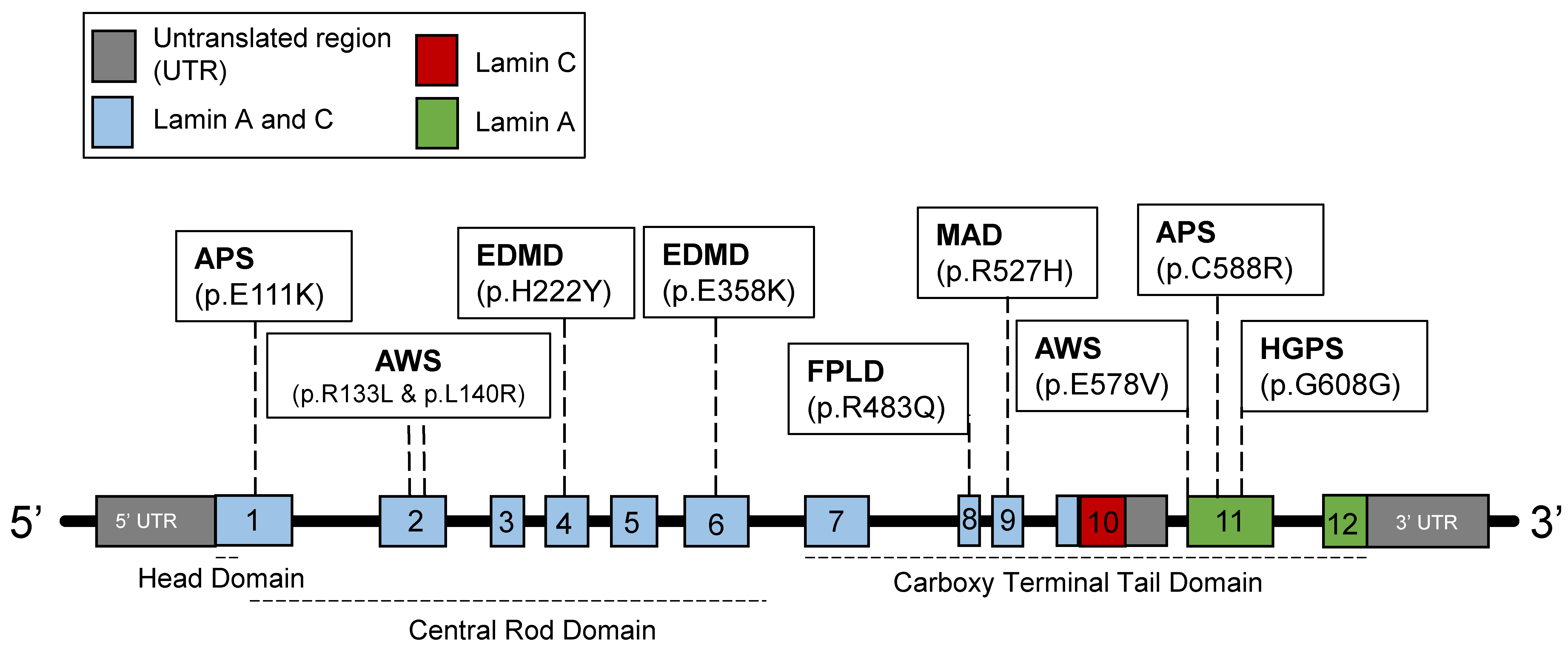
2.1. Introduction to Lamins and Laminopathies
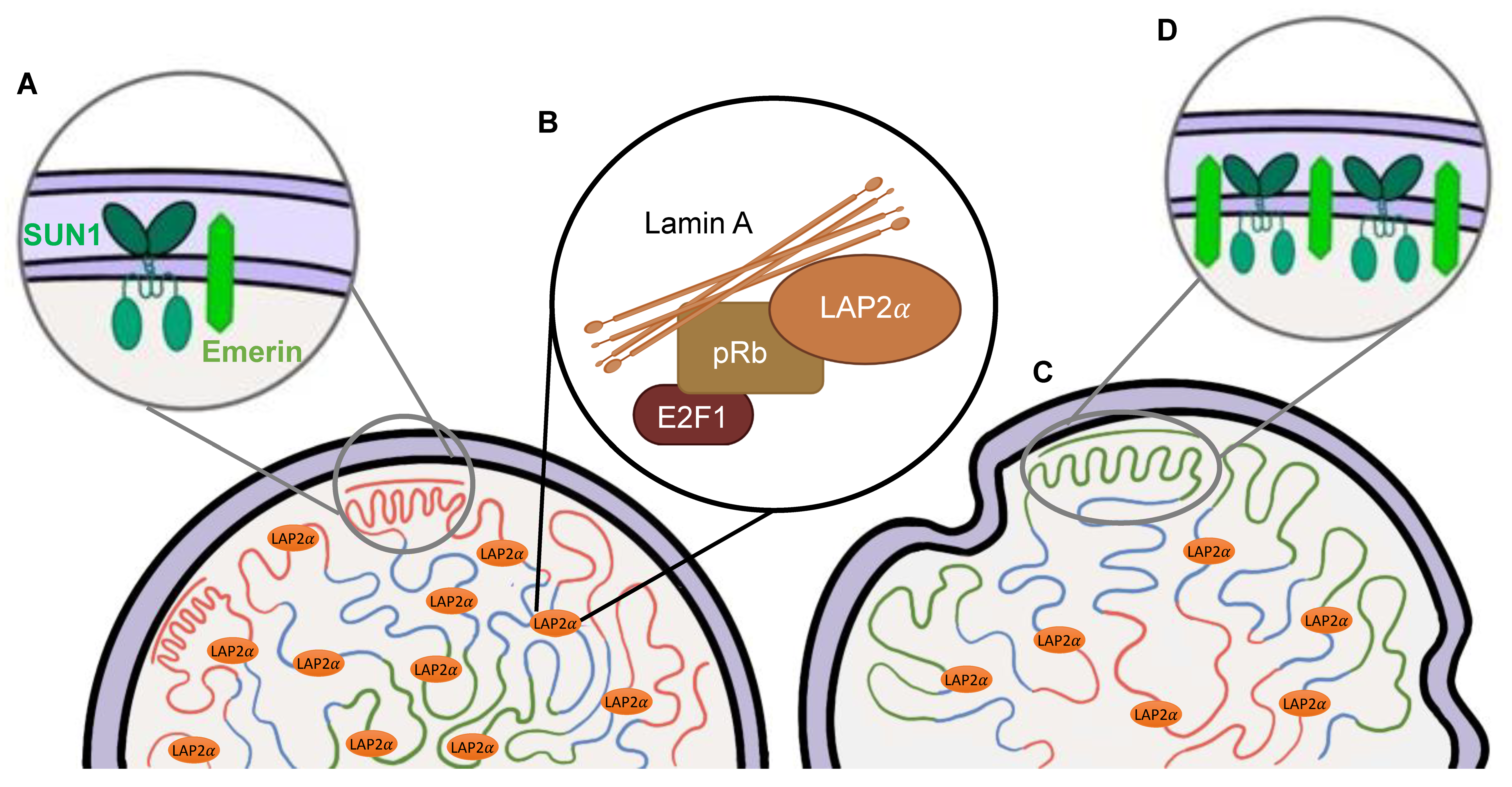
2.2. Physiological Functions of the Lamins
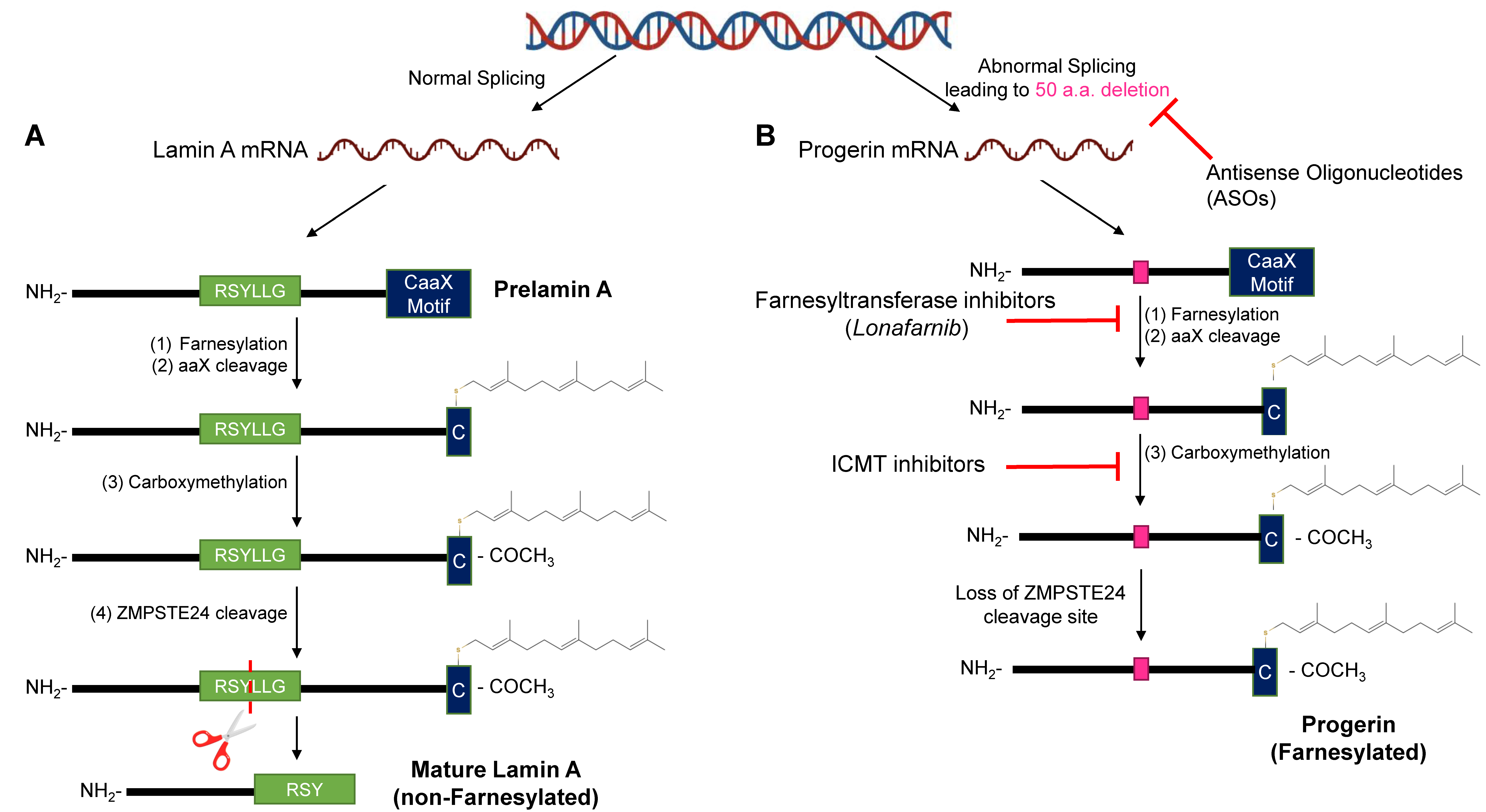
2.3. Disruptions of the Functions of Lamin A in HGPS
3. Progerin Expression Phenotypes
3.1. Nuclear Abnormalities
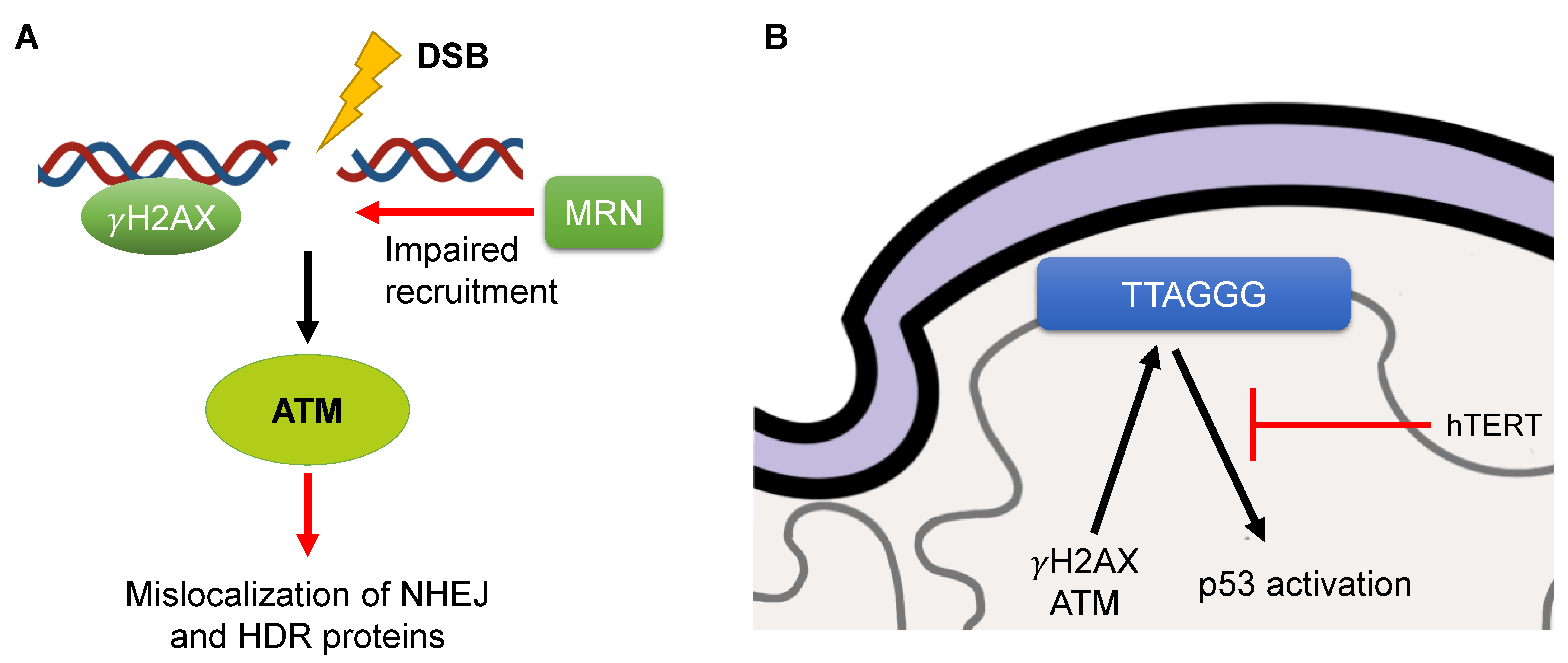
3.2. Defects in DNA Damage Response
3.3. Telomere Attrition
4. Tissue/Organ Dysfunction
5. Epigenetic Alterations in HGPS
5.1. Histone Modifications
5.2. DNA Methylation
5.3. LADs As a Potential Link
6. Treatments
6.1. Farnesyltransferase & GeranylGeranylTransferase Inhibitors
6.2. Isoprenylcysteine-Carboxyl-Methyltransferase Inhibitors
6.3. Antisense Oligonucleotides
6.4. CRISPR-Cas9 and Adenine Base Editors
6.5. Future Perspectives of Therapeutics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, F.; Kornak, U.; Wollnik, B. Premature aging disorders: A clinical and genetic compendium. Clin. Genet. 2021, 99, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J. Congenital Absence of Hair and Mammary Glands with Atrophic Condition of the Skin and its Appendages, in a Boy whose Mother had been almost wholly Bald from Alopecia Areata from the age of Six. Med.-Chir. Trans. 1886, 69, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65. [Google Scholar] [CrossRef]
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. [Google Scholar] [CrossRef]
- Janota, C.S.; Calero-Cuenca, F.J.; Gomes, E.R. The role of the cell nucleus in mechanotransduction. Curr. Opin. Cell Biol. 2020, 63, 204–211. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Lammerding, J. Novel insights into the disease etiology of laminopathies. Rare Dis. 2013, 1, e27002. [Google Scholar] [CrossRef]
- Rankin, J.; Ellard, S. The laminopathies: A clinical review. Clin. Genet. 2006, 70, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Helbling-Leclerc, A.; Bonne, G.; Schwartz, K. Emery-Dreifuss muscular dystrophy. Eur. J. Hum. Genet. 2002, 10, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Krawiec, P.; Melges, B.; Pac-Kozuchowska, E.; Mroczkowska-Juchkiewicz, A.; Czerska, K. Fitting the pieces of the puzzle together: A case report of the Dunnigan-type of familial partial lipodystrophy in the adolescent girl. BMC Pediatr. 2016, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef]
- Scharner, J.; Brown, C.A.; Bower, M.; Iannaccone, S.T.; Khatri, I.A.; Escolar, D.; Gordon, E.; Felice, K.; Crowe, C.A.; Grosmann, C.; et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum. Mutat. 2011, 32, 152–167. [Google Scholar] [CrossRef]
- Stierle, V.; Couprie, J.; Ostlund, C.; Krimm, I.; Zinn-Justin, S.; Hossenlopp, P.; Worman, H.J.; Courvalin, J.C.; Duband-Goulet, I. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 2003, 42, 4819–4828. [Google Scholar] [CrossRef]
- Chen, L.; Lee, L.; Kudlow, B.A.; Dos Santos, H.G.; Sletvold, O.; Shafeghati, Y.; Botha, E.G.; Garg, A.; Hanson, N.B.; Martin, G.M.; et al. LMNA mutations in atypical Werner’s syndrome. Lancet 2003, 362, 440–445. [Google Scholar] [CrossRef]
- Garg, A.; Subramanyam, L.; Agarwal, A.K.; Simha, V.; Levine, B.; D’Apice, M.R.; Novelli, G.; Crow, Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J. Clin. Endocrinol. Metab. 2009, 94, 4971–4983. [Google Scholar] [CrossRef]
- Seco-Cervera, M.; Spis, M.; Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Velazquez-Ledesma, A.; Esmoris, I.; Banuls, S.; Perez-Machado, G.; Pallardo, F.V. Oxidative stress and antioxidant response in fibroblasts from Werner and atypical Werner syndromes. Aging 2014, 6, 231–245. [Google Scholar] [CrossRef]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef]
- Dwyer, N.; Blobel, G. A modified procedure for the isolation of a pore complex-lamina fraction from rat liver nuclei. J. Cell Biol. 1976, 70, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Hosick, T.J.; Sinensky, M. Isoprenylation is required for the processing of the lamin A precursor. J. Cell Biol. 1990, 110, 1489–1499. [Google Scholar] [CrossRef]
- Casey, P.J.; Seabra, M.C. Protein prenyltransferases. J. Biol. Chem. 1996, 271, 5289–5292. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, D.P.; Kuszczak, D.; Rusinol, A.E.; Thewke, D.P.; Hrycyna, C.A.; Michaelis, S.; Sinensky, M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005, 387, 129–138. [Google Scholar] [CrossRef]
- Dai, Q.; Choy, E.; Chiu, V.; Romano, J.; Slivka, S.R.; Steitz, S.A.; Michaelis, S.; Philips, M.R. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J. Biol. Chem. 1998, 273, 15030–15034. [Google Scholar] [CrossRef]
- Sinensky, M.; Fantle, K.; Trujillo, M.; McLain, T.; Kupfer, A.; Dalton, M. The processing pathway of prelamin A. J. Cell Sci. 1994, 107 Pt 1, 61–67. [Google Scholar] [CrossRef]
- Weber, K.; Plessmann, U.; Traub, P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett. 1989, 257, 411–414. [Google Scholar] [CrossRef]
- Al-Saaidi, R.; Bross, P. Do lamin A and lamin C have unique roles? Chromosoma 2015, 124, 1–12. [Google Scholar] [CrossRef]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef]
- Broers, J.L.; Peeters, E.A.; Kuijpers, H.J.; Endert, J.; Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Ramaekers, F.C. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Hunt, C.R.; Pandita, R.K.; Kumar, R.; Yang, C.R.; Horikoshi, N.; Bachoo, R.; Serag, S.; Story, M.D.; Shay, J.W.; et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol. Cell. Biol. 2013, 33, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Spann, T.P.; Goldman, A.E.; Wang, C.; Huang, S.; Goldman, R.D. Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J. Cell Biol. 2002, 156, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Korbei, B.; Vaughan, O.A.; Vlcek, S.; Hutchison, C.J.; Foisner, R. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J. Cell Sci. 2000, 113 Pt 19, 3473–3484. [Google Scholar] [CrossRef]
- Elenbaas, J.S.; Bragazzi Cunha, J.; Azuero-Dajud, R.; Nelson, B.; Oral, E.A.; Williams, J.A.; Stewart, C.L.; Omary, M.B. Lamin A/C Maintains Exocrine Pancreas Homeostasis by Regulating Stability of RB and Activity of E2F. Gastroenterology 2018, 154, 1625–1629 e1628. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Legartova, S.; Fagherazzi, P.; Stixova, L.; Kovarik, A.; Raska, I.; Bartova, E. The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction. Cells 2021, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Keijzers, G.; Akbari, M.; Ezra, M.B.; Hall, A.; Morevati, M.; Scheibye-Knudsen, M.; Gonzalo, S.; Bartek, J.; Bohr, V.A. Lamin A/C promotes DNA base excision repair. Nucleic Acids Res. 2019, 47, 11709–11728. [Google Scholar] [CrossRef]
- Rodriguez, J.; Calvo, F.; Gonzalez, J.M.; Casar, B.; Andres, V.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Hall, A.; Galanos, P.; Rizza, S.; Yamamoto, T.; Gram, H.H.; Munk, S.H.N.; Shoaib, M.; Sorensen, C.S.; Bohr, V.A.; et al. Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1alpha and the NAMPT-NAD+ pathway. Nucleic Acids Res. 2022, 50, 9948–9965. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hu, J.; Yue, S.; Kristiani, L.; Kim, M.; Sauria, M.; Taylor, J.; Kim, Y.; Zheng, Y. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell 2018, 71, 802–815 e807. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- Decker, M.L.; Chavez, E.; Vulto, I.; Lansdorp, P.M. Telomere length in Hutchinson-Gilford progeria syndrome. Mech. Ageing Dev. 2009, 130, 377–383. [Google Scholar] [CrossRef]
- Liu, Y.; Rusinol, A.; Sinensky, M.; Wang, Y.; Zou, Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J. Cell Sci. 2006, 119, 4644–4649. [Google Scholar] [CrossRef]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef]
- Columbaro, M.; Capanni, C.; Mattioli, E.; Novelli, G.; Parnaik, V.K.; Squarzoni, S.; Maraldi, N.M.; Lattanzi, G. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. Life Sci. 2005, 62, 2669–2678. [Google Scholar] [CrossRef]
- Fan, Q.; Li, X.M.; Zhai, C.; Li, B.; Li, S.T.; Dong, M.Q. Somatic nuclear blebbing in Caenorhabditis elegans is not a feature of organismal aging but a potential indicator of germline proliferation in early adulthood. G3 2023, jkad029. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.H.; Chen, N.Y.; Heizer, P.J.; Tu, Y.; Weston, T.A.; Fong, J.L.; Gill, N.K.; Rowat, A.C.; Young, S.G.; Fong, L.G. Nuclear membrane ruptures underlie the vascular pathology in a mouse model of Hutchinson-Gilford progeria syndrome. JCI Insight 2021, 6, e151515. [Google Scholar] [CrossRef]
- Pegoraro, G.; Kubben, N.; Wickert, U.; Gohler, H.; Hoffmann, K.; Misteli, T. Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 2009, 11, 1261–1267. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Wang, W.P.; Chen, Y.C.; Wang, J.Y.; Lin, W.H.; Tai, L.A.; Liou, G.G.; Yang, C.S.; Chi, Y.H. Dysregulated interactions between lamin A and SUN1 induce abnormalities in the nuclear envelope and endoplasmic reticulum in progeric laminopathies. J. Cell Sci. 2014, 127, 1792–1804. [Google Scholar] [CrossRef]
- Arii, J.; Maeda, F.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Mori, Y.; Kawaguchi, Y. ESCRT-III controls nuclear envelope deformation induced by progerin. Sci. Rep. 2020, 10, 18877. [Google Scholar] [CrossRef] [PubMed]
- Booth, E.A.; Spagnol, S.T.; Alcoser, T.A.; Dahl, K.N. Nuclear stiffening and chromatin softening with progerin expression leads to an attenuated nuclear response to force. Soft Matter 2015, 11, 6412–6418. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Ji, J.Y.; Cummings, K.S.; Lee, R.T.; Lammerding, J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: Effects of farnesyltransferase inhibitors. Aging Cell 2008, 7, 383–393. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Tonoyama, Y.; Shinya, M.; Toyoda, A.; Kitano, T.; Oga, A.; Nishimaki, T.; Katsumura, T.; Oota, H.; Wan, M.T.; Yip, B.W.P.; et al. Abnormal nuclear morphology is independent of longevity in a zmpste24-deficient fish model of Hutchinson-Gilford progeria syndrome (HGPS). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2018, 209, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935 e925. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Milholland, B.; de Bruin, A.; Curran, S.; Laberge, R.M.; van Steeg, H.; Campisi, J.; Maslov, A.Y.; Vijg, J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun. 2015, 6, 6790. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.D.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Rusinol, A.E.; Sinensky, M.S.; Liu, J.; Shell, S.M.; Zou, Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J. 2008, 22, 603–611. [Google Scholar] [CrossRef]
- Manju, K.; Muralikrishna, B.; Parnaik, V.K. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J. Cell Sci. 2006, 119, 2704–2714. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Z.; Ghosh, S.; Zhou, Z. Defective ATM-Kap-1-mediated chromatin remodeling impairs DNA repair and accelerates senescence in progeria mouse model. Aging Cell 2013, 12, 316–318. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Zhang, H.; Xiong, Z.M.; Cao, K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. USA 2014, 111, E2261–E2270. [Google Scholar] [CrossRef]
- Wheaton, K.; Campuzano, D.; Ma, W.; Sheinis, M.; Ho, B.; Brown, G.W.; Benchimol, S. Progerin-Induced Replication Stress Facilitates Premature Senescence in Hutchinson-Gilford Progeria Syndrome. Mol. Cell. Biol. 2017, 37, e00659-16. [Google Scholar] [CrossRef]
- Constantinescu, D.; Csoka, A.B.; Navara, C.S.; Schatten, G.P. Defective DSB repair correlates with abnormal nuclear morphology and is improved with FTI treatment in Hutchinson-Gilford progeria syndrome fibroblasts. Exp. Cell Res. 2010, 316, 2747–2759. [Google Scholar] [CrossRef]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef]
- Meyer, J.N.; Boyd, W.A.; Azzam, G.A.; Haugen, A.C.; Freedman, J.H.; Van Houten, B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol. 2007, 8, R70. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, L.; Zhang, W.; Meng, D.; Zhang, H.; Jiang, Y.; Xu, X.; Van Meter, M.; Seluanov, A.; Gorbunova, V.; et al. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 14, 269–276. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Aguado, J.; Sola-Carvajal, A.; Cancila, V.; Revechon, G.; Ong, P.F.; Jones-Weinert, C.W.; Wallen Arzt, E.; Lattanzi, G.; Dreesen, O.; Tripodo, C.; et al. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson-Gilford Progeria Syndrome. Nat. Commun. 2019, 10, 4990. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.; Lim, J.S.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2alpha-telomere association in Hutchinson-Gilford progeria. Elife 2015, 4, e07759. [Google Scholar] [CrossRef]
- Lin, H.; Mensch, J.; Haschke, M.; Jager, K.; Kottgen, B.; Dernedde, J.; Orso, E.; Walter, M. Establishment and Characterization of hTERT Immortalized Hutchinson-Gilford Progeria Fibroblast Cell Lines. Cells 2022, 11, 2784. [Google Scholar] [CrossRef]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Investig. 2011, 121, 2833–2844. [Google Scholar] [CrossRef]
- De Vos, W.H.; Houben, F.; Hoebe, R.A.; Hennekam, R.; van Engelen, B.; Manders, E.M.; Ramaekers, F.C.; Broers, J.L.; Van Oostveldt, P. Increased plasticity of the nuclear envelope and hypermobility of telomeres due to the loss of A-type lamins. Biochim. Biophys. Acta 2010, 1800, 448–458. [Google Scholar] [CrossRef]
- Fernandez, P.; Scaffidi, P.; Markert, E.; Lee, J.H.; Rane, S.; Misteli, T. Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep. 2014, 9, 248–260. [Google Scholar] [CrossRef]
- Sarkar, P.K.; Shinton, R.A. Hutchinson-Guilford progeria syndrome. Postgrad. Med. J. 2001, 77, 312–317. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W.; Progeria Clinical Trials, C. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef]
- Ullrich, N.J.; Silvera, V.M.; Campbell, S.E.; Gordon, L.B. Craniofacial abnormalities in Hutchinson-Gilford progeria syndrome. AJNR Am. J. Neuroradiol. 2012, 33, 1512–1518. [Google Scholar] [CrossRef]
- Gordon, L.B.; McCarten, K.M.; Giobbie-Hurder, A.; Machan, J.T.; Campbell, S.E.; Berns, S.D.; Kieran, M.W. Disease progression in Hutchinson-Gilford progeria syndrome: Impact on growth and development. Pediatrics 2007, 120, 824–833. [Google Scholar] [CrossRef]
- Papadogkonas, G.; Papamatheakis, D.A.; Spilianakis, C. 3D Genome Organization as an Epigenetic Determinant of Transcription Regulation in T Cells. Front. Immunol. 2022, 13, 921375. [Google Scholar] [CrossRef]
- Wang, J.; Huang, T.Y.; Hou, Y.; Bartom, E.; Lu, X.; Shilatifard, A.; Yue, F.; Saratsis, A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 2021, 7, eabg4126. [Google Scholar] [CrossRef]
- Kohler, F.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Corless, S.; Musch, T.; Lonsdorf, A.S.; Erhardt, S.; Lyko, F.; Rodriguez-Paredes, M. Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome. Genome Med. 2020, 12, 46. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef]
- Villeponteau, B. The heterochromatin loss model of aging. Exp. Gerontol. 1997, 32, 383–394. [Google Scholar] [CrossRef]
- Cao, K.; Capell, B.C.; Erdos, M.R.; Djabali, K.; Collins, F.S. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4949–4954. [Google Scholar] [CrossRef]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 2013, 4, 1868. [Google Scholar] [CrossRef]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Foo, M.X.R.; Liebl, D.; Hor, L.P.; Stewart, C.L.; Dreesen, O. Heterochromatin loss as a determinant of progerin-induced DNA damage in Hutchinson-Gilford Progeria. Aging Cell 2020, 19, e13108. [Google Scholar] [CrossRef]
- Heyn, H.; Moran, S.; Esteller, M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics 2013, 8, 28–33. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef]
- Sebestyen, E.; Marullo, F.; Lucini, F.; Petrini, C.; Bianchi, A.; Valsoni, S.; Olivieri, I.; Antonelli, L.; Gregoretti, F.; Oliva, G.; et al. SAMMY-seq reveals early alteration of heterochromatin and deregulation of bivalent genes in Hutchinson-Gilford Progeria Syndrome. Nat. Commun. 2020, 11, 6274. [Google Scholar] [CrossRef]
- Manzo, S.G.; Dauban, L.; van Steensel, B. Lamina-associated domains: Tethers and looseners. Curr. Opin. Cell Biol. 2022, 74, 80–87. [Google Scholar] [CrossRef]
- Bejaoui, Y.; Razzaq, A.; Yousri, N.A.; Oshima, J.; Megarbane, A.; Qannan, A.; Potabattula, R.; Alam, T.; Martin, G.M.; Horn, H.F.; et al. DNA methylation signatures in Blood DNA of Hutchinson-Gilford Progeria syndrome. Aging Cell 2022, 21, e13555. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Graf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic Reorganization of Lamin-Associated Domains in Cardiac Myocytes Is Associated with Differential Gene Expression and DNA Methylation in Human Dilated Cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.G.; Frost, D.; Meta, M.; Qiao, X.; Yang, S.H.; Coffinier, C.; Young, S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006, 311, 1621–1623. [Google Scholar] [CrossRef]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- Agrawal, A.G.; Somani, R.R. Farnesyltransferase inhibitor as anticancer agent. Mini Rev. Med. Chem. 2009, 9, 638–652. [Google Scholar] [CrossRef]
- Bishop, W.R.; Bond, R.; Petrin, J.; Wang, L.; Patton, R.; Doll, R.; Njoroge, G.; Catino, J.; Schwartz, J.; Windsor, W.; et al. Novel tricyclic inhibitors of farnesyl protein transferase. Biochemical characterization and inhibition of Ras modification in transfected Cos cells. J. Biol. Chem. 1995, 270, 30611–30618. [Google Scholar] [CrossRef]
- Moores, S.L.; Schaber, M.D.; Mosser, S.D.; Rands, E.; O’Hara, M.B.; Garsky, V.M.; Marshall, M.S.; Pompliano, D.L.; Gibbs, J.B. Sequence dependence of protein isoprenylation. J. Biol. Chem. 1991, 266, 14603–14610. [Google Scholar] [CrossRef]
- Reiss, Y.; Goldstein, J.L.; Seabra, M.C.; Casey, P.J.; Brown, M.S. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell 1990, 62, 81–88. [Google Scholar] [CrossRef]
- Lee, S.J.; Jung, Y.S.; Yoon, M.H.; Kang, S.M.; Oh, A.Y.; Lee, J.H.; Jun, S.Y.; Woo, T.G.; Chun, H.Y.; Kim, S.K.; et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Investig. 2016, 126, 3879–3893. [Google Scholar] [CrossRef]
- Wang, Y.; Ostlund, C.; Choi, J.C.; Swayne, T.C.; Gundersen, G.G.; Worman, H.J. Blocking farnesylation of the prelamin A variant in Hutchinson-Gilford progeria syndrome alters the distribution of A-type lamins. Nucleus 2012, 3, 452–462. [Google Scholar] [CrossRef]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 2005, 102, 12873–12878. [Google Scholar] [CrossRef]
- Bikkul, M.U.; Clements, C.S.; Godwin, L.S.; Goldberg, M.W.; Kill, I.R.; Bridger, J.M. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology 2018, 19, 579–602. [Google Scholar] [CrossRef]
- Windmueller, R.; Leach, J.P.; Babu, A.; Zhou, S.; Morley, M.P.; Wakabayashi, A.; Petrenko, N.B.; Viatour, P.; Morrisey, E.E. Direct Comparison of Mononucleated and Binucleated Cardiomyocytes Reveals Molecular Mechanisms Underlying Distinct Proliferative Competencies. Cell Rep. 2020, 30, 3105–3116.e3104. [Google Scholar] [CrossRef]
- Capell, B.C.; Olive, M.; Erdos, M.R.; Cao, K.; Faddah, D.A.; Tavarez, U.L.; Conneely, K.N.; Qu, X.; San, H.; Ganesh, S.K.; et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 15902–15907. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children with Hutchinson-Gilford Progeria Syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.L.; Suarez, M.F.; Cau, P.; Cadinanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef]
- Cubria, M.B.; Suarez, S.; Masoudi, A.; Oftadeh, R.; Kamalapathy, P.; DuBose, A.; Erdos, M.R.; Cabral, W.A.; Karim, L.; Collins, F.S.; et al. Evaluation of musculoskeletal phenotype of the G608G progeria mouse model with lonafarnib, pravastatin, and zoledronic acid as treatment groups. Proc. Natl. Acad. Sci. USA 2020, 117, 12029–12040. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 2011, 108, 4997–5002. [Google Scholar] [CrossRef]
- Phase I/II Trial of Everolimus in Combination with Lonafarnib in Progeria. Available online: https://clinicaltrials.gov/ct2/show/NCT02579044 (accessed on 7 December 2022).
- Arnold, R.; Vehns, E.; Randl, H.; Djabali, K. Baricitinib, a JAK-STAT Inhibitor, Reduces the Cellular Toxicity of the Farnesyltransferase Inhibitor Lonafarnib in Progeria Cells. Int. J. Mol. Sci. 2021, 22, 7474. [Google Scholar] [CrossRef]
- Davies, B.S.; Barnes, R.H., 2nd; Tu, Y.; Ren, S.; Andres, D.A.; Spielmann, H.P.; Lammerding, J.; Wang, Y.; Young, S.G.; Fong, L.G. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum. Mol. Genet. 2010, 19, 2682–2694. [Google Scholar] [CrossRef]
- Almendariz-Palacios, C.; Gillespie, Z.E.; Janzen, M.; Martinez, V.; Bridger, J.M.; Harkness, T.A.A.; Mousseau, D.D.; Eskiw, C.H. The Nuclear Lamina: Protein Accumulation and Disease. Biomedicines 2020, 8, 188. [Google Scholar] [CrossRef]
- Davies, B.S.; Fong, L.G.; Yang, S.H.; Coffinier, C.; Young, S.G. The posttranslational processing of prelamin A and disease. Annu. Rev. Genom. Hum. Genet. 2009, 10, 153–174. [Google Scholar] [CrossRef]
- Chen, X.; Yao, H.; Kashif, M.; Revechon, G.; Eriksson, M.; Hu, J.; Wang, T.; Liu, Y.; Tuksammel, E.; Stromblad, S.; et al. A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. Elife 2021, 10, e63284. [Google Scholar] [CrossRef]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef]
- Marcos-Ramiro, B.; Gil-Ordonez, A.; Marin-Ramos, N.I.; Ortega-Nogales, F.J.; Balabasquer, M.; Gonzalo, P.; Khiar-Fernandez, N.; Rolas, L.; Barkaway, A.; Nourshargh, S.; et al. Isoprenylcysteine Carboxylmethyltransferase-Based Therapy for Hutchinson-Gilford Progeria Syndrome. ACS Cent. Sci. 2021, 7, 1300–1310. [Google Scholar] [CrossRef]
- Fong, L.G.; Vickers, T.A.; Farber, E.A.; Choi, C.; Yun, U.J.; Hu, Y.; Yang, S.H.; Coffinier, C.; Lee, R.; Yin, L.; et al. Activating the synthesis of progerin, the mutant prelamin A in Hutchinson-Gilford progeria syndrome, with antisense oligonucleotides. Hum. Mol. Genet. 2009, 18, 2462–2471. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Levy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef]
- Erdos, M.R.; Cabral, W.A.; Tavarez, U.L.; Cao, K.; Gvozdenovic-Jeremic, J.; Narisu, N.; Zerfas, P.M.; Crumley, S.; Boku, Y.; Hanson, G.; et al. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 536–545. [Google Scholar] [CrossRef]
- Puttaraju, M.; Jackson, M.; Klein, S.; Shilo, A.; Bennett, C.F.; Gordon, L.; Rigo, F.; Misteli, T. Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 526–535. [Google Scholar] [CrossRef]
- Beyret, E.; Liao, H.K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G.; Reddy, P.; Izpisua Belmonte, J.C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef]
- Santiago-Fernandez, O.; Osorio, F.G.; Quesada, V.; Rodriguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef]
- Liu, G.H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Whisenant, D.; Lim, K.; Revechon, G.; Yao, H.; Bergo, M.O.; Machtel, P.; Kim, J.S.; Eriksson, M. Transient expression of an adenine base editor corrects the Hutchinson-Gilford progeria syndrome mutation and improves the skin phenotype in mice. Nat. Commun. 2022, 13, 3068. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Ahn, J.; Kim, J.E.; Kim, S.Y.; Kang, S.Y.; Joo, J.; Park, S.; Cho, J.H.; Woo, T.G.; et al. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun. Biol. 2021, 4, 5. [Google Scholar] [CrossRef]
- Vehns, E.; Arnold, R.; Djabali, K. Impact of MnTBAP and Baricitinib Treatment on Hutchinson-Gilford Progeria Fibroblasts. Pharmaceuticals 2022, 15, 945. [Google Scholar] [CrossRef]
- Mojiri, A.; Walther, B.K.; Jiang, C.; Matrone, G.; Holgate, R.; Xu, Q.; Morales, E.; Wang, G.; Gu, J.; Wang, R.; et al. Telomerase therapy reverses vascular senescence and extends lifespan in progeria mice. Eur. Heart J. 2021, 42, 4352–4369. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Qin, W.; Tang, X.; Meng, Y.; Hu, W.; Zhang, S.; Qian, M.; Liu, Z.; Cao, X.; Pang, Q.; et al. Vascular endothelium-targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci. Adv. 2020, 6, eaay5556. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista, N.J.; Desai, S.G.; Perez, A.M.; Finkelstein, A.; Radigan, R.; Singh, M.; Landman, A.; Drittel, B.; Abramov, D.; Ahsan, M.; et al. The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments. Genes 2023, 14, 602. https://doi.org/10.3390/genes14030602
Batista NJ, Desai SG, Perez AM, Finkelstein A, Radigan R, Singh M, Landman A, Drittel B, Abramov D, Ahsan M, et al. The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments. Genes. 2023; 14(3):602. https://doi.org/10.3390/genes14030602
Chicago/Turabian StyleBatista, Noelle J., Sanket G. Desai, Alexis M. Perez, Alexa Finkelstein, Rachel Radigan, Manrose Singh, Aaron Landman, Brian Drittel, Daniella Abramov, Mina Ahsan, and et al. 2023. "The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments" Genes 14, no. 3: 602. https://doi.org/10.3390/genes14030602
APA StyleBatista, N. J., Desai, S. G., Perez, A. M., Finkelstein, A., Radigan, R., Singh, M., Landman, A., Drittel, B., Abramov, D., Ahsan, M., Cornwell, S., & Zhang, D. (2023). The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments. Genes, 14(3), 602. https://doi.org/10.3390/genes14030602