
De Novo Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Chaenomeles speciosa (Sweet) Nakai Revealed the Existence of Two Structural Isomers

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequencing
2.2. Assembly and Annotation of the Mitochondrial Genome and Chloroplast Genome
2.3. Analysis of Repeated Sequences and Genome Recombination
2.4. DNA Transfer and Phylogenetic Tree Construction
2.5. RNA Editing Site Identification and Synteny Analysis
2.6. dN/dS Analysis
3. Results
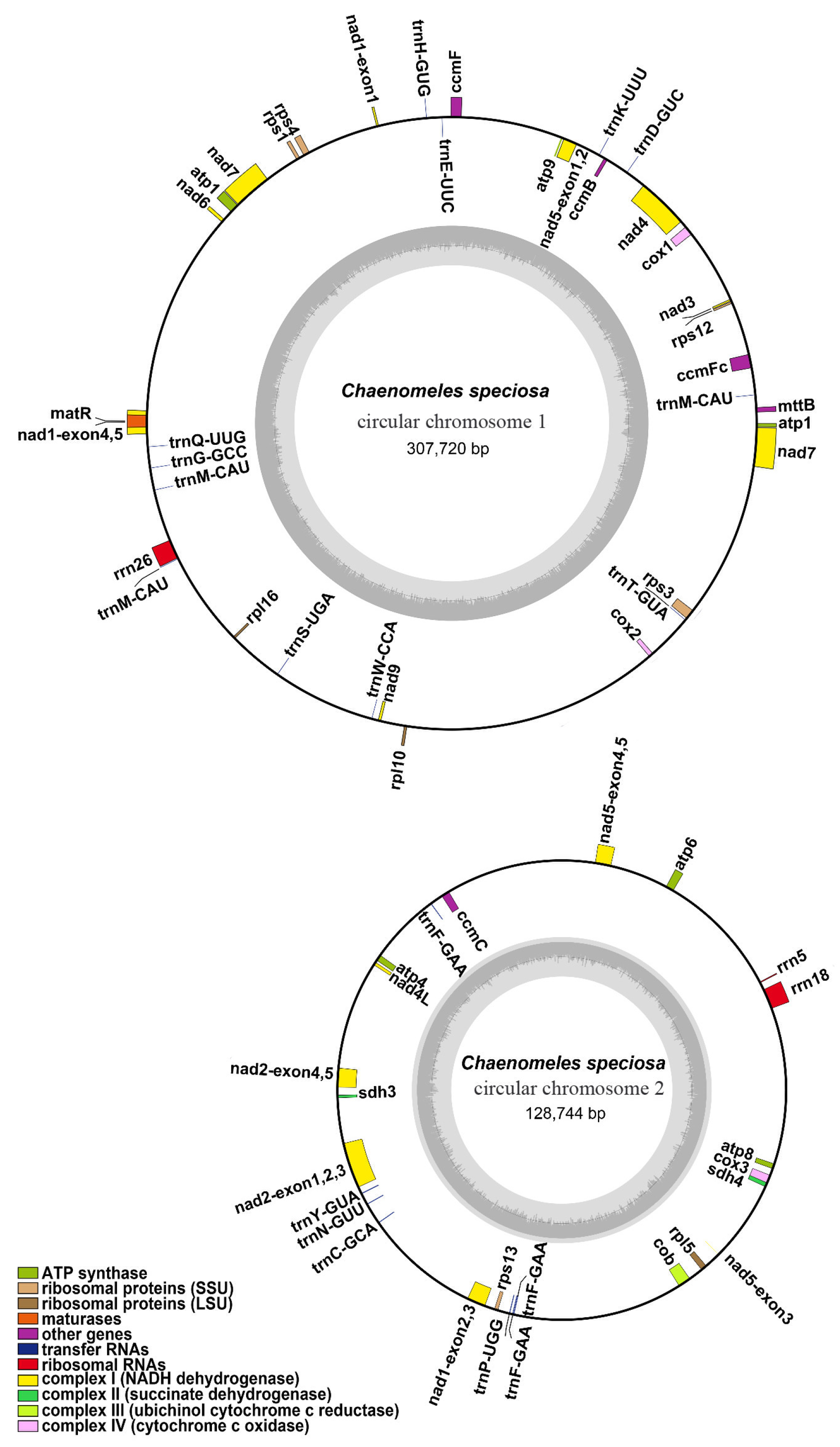
3.1. General Features of the C. speciosa Mitochondrial Genome
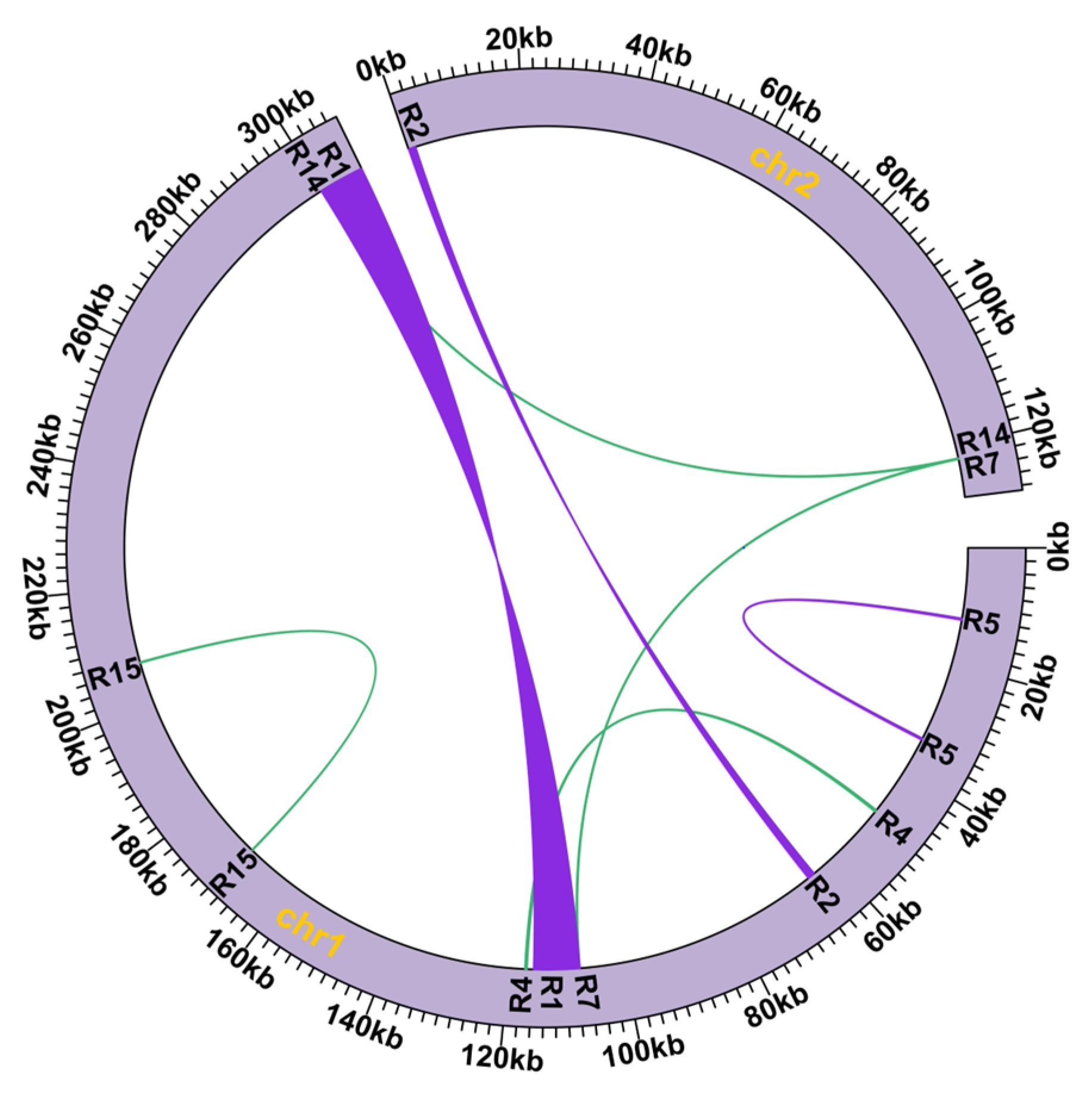
3.2. Repeats and Homologous Recombination
3.3. Gene Transfers between Chloroplast and Mitochondrial Genomes
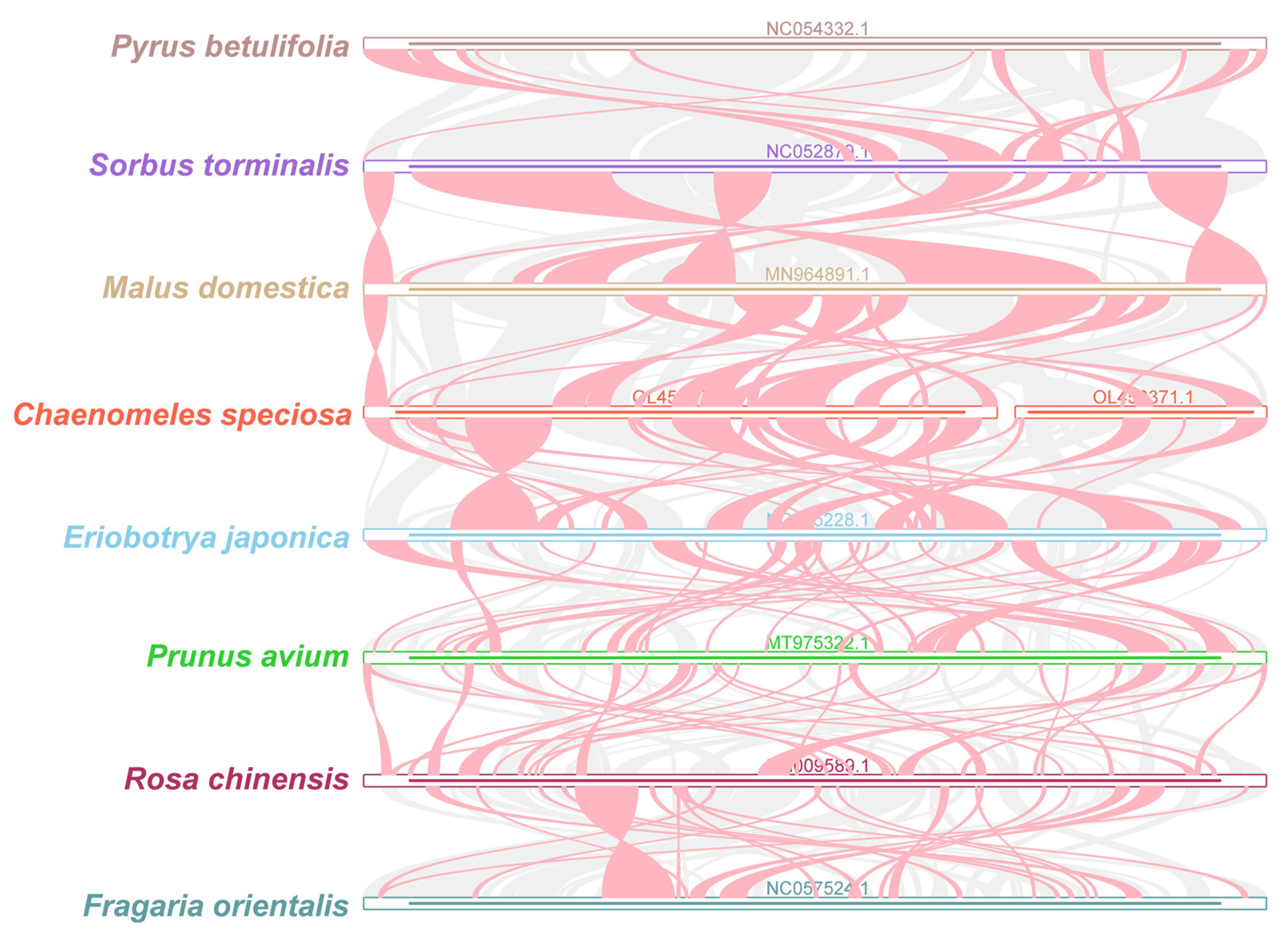
3.4. Phylogenetic Analysis
3.5. RNA Editing Sites Prediction
3.6. Synteny Analysis
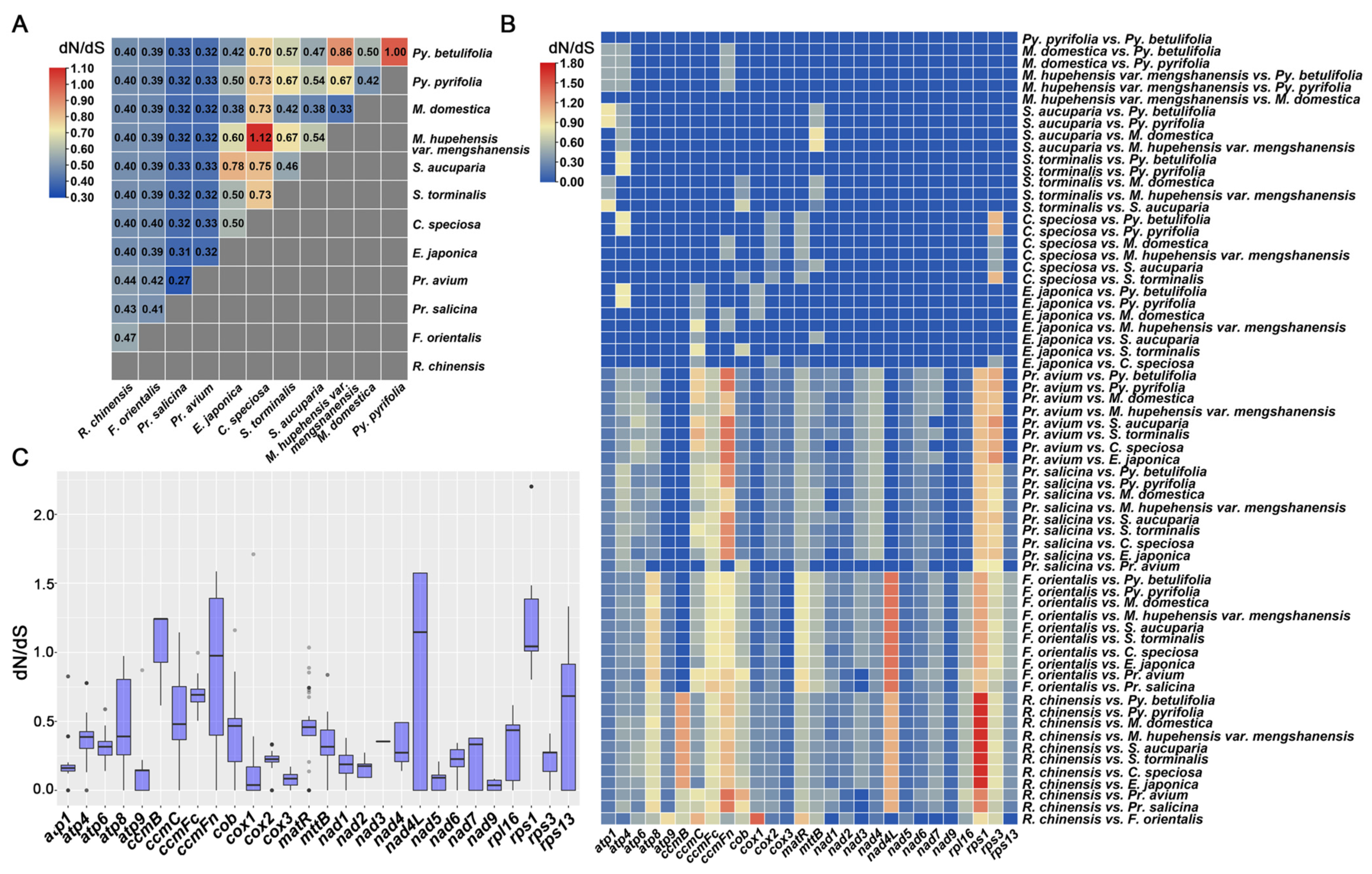
3.7. Evolutionary Selection Pressure Analysis
4. Discussion
4.1. Features of the C. speciosa Mitochondrial Genome and IGT Events
4.2. The Conformations of C. speciosa Mitochondrial Genome Mediated by Repeated Sequences
4.3. Evolutionary Analysis and dN/dS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Yang, Y.-B.; Yang, Q.; Sun, L.-N.; Chen, W.-S.; Park, Y.-S.; Towantakavanit, K.; Kowalska, T.; Jung, S.-T.; Ham, K.-S.; et al. Anti-Inflammatory and Analgesic Activities of Chaenomeles speciosa Fractions in Laboratory Animals. J. Med. Food 2009, 12, 1016–1022. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Y.X.; Liu, A.L.; Wang, H.D.; Wang, Y.L.; Du, G.H. Antioxidant, anti-inflammatory and anti-influenza properties of components from Chaenomeles Speciosa. Molecules 2010, 15, 8507–8517. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Xu, H.Y.; He, H.B.; Li, X.M.; Feng, M.L.; He, Y.M.; Jiang, W.J.; Wang, J.Z.; Xu, D.X.; Zou, K. Total triterpenes from the fruits of Chaenomeles speciosa (Sweet) Nakai protects against indomethacin-induced gastric mucosal injury: Involvement of TFF1-mediated EGF/EGFR and apoptotic pathways. J. Pharm. Pharm. 2020, 72, 409–423. [Google Scholar] [CrossRef]
- Ma, Y.B.; Li, J.H.; Li, J.L.; Yang, L.; Wu, J.L.; Liu, S.Y. Comparative Metabolomics Study of Chaenomeles speciosa (Sweet) Nakai from Different Geographical Regions. Foods 2022, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-H.; Xi, Z.-X.; Li, J.L.; Chen, C.; Yang, G.J.; Sun, L.N.; Chen, W.S.; Zhu, H.Y. Isolation of two new phenolic compounds from the fruit of Chaenomeles speciosa (Sweet) Nakai. Phytochem. Lett. 2013, 6, 526–530. [Google Scholar] [CrossRef]
- Miao, J.; Li, X.; Zhao, C.C.; Gao, X.X.; Wang, Y.; Gao, W.Y. Active compounds, antioxidant activity and α-glucosidase inhibitory activity of different varieties of Chaenomeles fruits. Food Chem. 2018, 248, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef]
- Birky, C.W., Jr. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef]
- Birky, C.W., Jr. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Mackenzie, S.; McIntosh, L. Higher plant mitochondria. Plant Cell 1999, 11, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Zhuang, Y.; Zhang, P.; Adams, K.L. Comparative Analysis of Structural Diversity and Sequence Evolution in Plant Mitochondrial Genes Transferred to the Nucleus. Mol. Biol. Evol. 2009, 26, 16. [Google Scholar] [CrossRef]
- Touzet, P.; Meyer, E.H. Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion 2014, 19 Pt B, 166–171. [Google Scholar] [CrossRef]
- Christensen, A.C. Mitochondrial DNA Repair and Genome Evolution. Annu. Plant Rev. 2018, 50, 11–32. [Google Scholar]
- Stewart, A.; Morley, B.L.N. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Lin, Q.; Banerjee, A.; Stefanovic, S. Mitochondrial phylogenomics of Cuscuta (Convolvulaceae) reveals a potentially functional horizontal gene transfer from the host. Genome Biol. Evol. 2022, 14, evac091. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, S. Complete Plastid and Mitochondrial Genomes of Aeginetia indica Reveal Intracellular Gene Transfer (IGT), Horizontal Gene Transfer (HGT), and Cytoplasmic Male Sterility (CMS). Int. J. Mol. Sci. 2021, 22, 6143. [Google Scholar] [CrossRef]
- Anderson, B.M.; Krause, K.; Petersen, G. Mitochondrial genomes of two parasitic Cuscuta species lack clear evidence of horizontal gene transfer and retain unusually fragmented ccmFC genes. BMC Genom. 2021, 22, 816. [Google Scholar] [CrossRef]
- Park, S.; Grewe, F.; Zhu, A.D.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Keeling, P.J. Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. USA 2015, 112, 10177–10184. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multi-chromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef]
- Liu, D.; Guo, H.L.; Zhu, J.L.; Qu, K.; Chen, Y.; Guo, Y.T.; Ding, P.; Yang, H.P.; Xu, T.; Jing, Q.; et al. Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant. Genes 2022, 13, 1321. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puerta, M.V.; Garcia, L.E.; Wohlfeiler, J.; Ceriotti, L.F. Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytol. 2017, 214, 376–387. [Google Scholar] [CrossRef]
- Yu, R.; Sun, C.; Zhong, Y.; Liu, Y.; Sanchez-Puerta, M.V.; Mower, J.P.; Zhou, R.C. The minicircular and extremely heteroplasmic mitogenome of the holoparasitic plant Rhopalocnemis phalloides. Curr. Biol. 2022, 32, 470–479.e475. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant. Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Mower, J.P.; Case, A.L.; Floro, E.R.; Willis, J.H. Evidence against Equimolarity of Large Repeat Arrangements and a Predominant Master Circle Structure of the Mitochondrial Genome from a Monkeyflower (Mimulus guttatus) Lineage with Cryptic CMS. Genome Biol. Evol. 2012, 4, 670–686. [Google Scholar] [CrossRef]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence, and Evolution. G3 Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Chevigny, N.; Schatz-Daas, D.; Lotfi, F.; Gualberto, J.M. DNA Repair and the Stability of the Plant Mitochondrial Genome. Int. J. Mol. Sci. 2020, 21, 328. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Dong, S.; Liu, Y.; Liao, W.; Zhang, S. The complete mitochondrial genome of Cycas debaoensis revealed unexpected static evolution in gymnosperm species. PLoS One 2021, 16, e0255091. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Liao, X.Z.; Zhang, X.N.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multi-chromosomal structuring. J. Syst. Evol. 2020, 60, 160–168. [Google Scholar] [CrossRef]
- Sloan, D.B.; Wu, Z. Molecular Evolution: The Perplexing Diversity of Mitochondrial RNA Editing Systems. Curr. Biol. 2016, 26, 22–24. [Google Scholar] [CrossRef]
- Morley, S.A.; Ahmad, N.; Nielsen, B.L. Plant Organelle Genome Replication. Plants 2019, 8, 358. [Google Scholar] [CrossRef]
- Sun, J.H.; Wang, Y.H.; Liu, Y.L.; Xu, C.; Yuan, Q.J.; Guo, L.P.; Huang, L.Q. Evolutionary and phylogenetic aspects of the chloroplast genome of Chaenomeles species. Sci. Rep. 2020, 10, 11466. [Google Scholar] [CrossRef]
- Mavrodiev, E.V.; Dervinis, C. A new, simple, highly scalable, and efficient protocol for genomic DNA extraction from diverse plant taxa. Appl. Plant Sci. 2021, 9, e11413. [Google Scholar] [CrossRef]
- Emerman, A.B.; Bowman, S.K.; Barry, A.; Henig, A.; Patel, K.M.; Gardner, A.F.; Hendrickson, C.L. NEBNext Direct: A Novel, Rapid, Hybridization-Based Approach for the Capture and Library Conversion of Genomic Regions of Interest. Curr. Protoc. Mol. Biol. 2017, 119, 7.30.31–37.30.24. [Google Scholar] [CrossRef]
- Wright, M.N.; Gola, D.; Ziegler, A. Preprocessing and Quality Control for Whole-Genome Sequences from the Illumina HiSeq X Platform. Methods Mol Biol 2017, 1666, 629–647. [Google Scholar] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–w73. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.L.; Zhang, X.Y.; Yang, H.Y.; Chen, H.M.; Liu, C. CPGVIEW: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 00, 1–11. [Google Scholar]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.M.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. Bui QM: IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Alexandros, S. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar]
- Huelsenbeck, J.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Lenz, H.; Hein, A.; Knoop, V. Plant organelle RNA editing and its specificity factors: Enhancements of analyses and new database features in PREPACT 3. 0. BMC Bioinform. 2018, 19, 255. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.-H.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis; Taylor & Francis: New York, NY, USA, 2019. [Google Scholar]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, M.; Chen, X.N.; Liu, Y.Y.; Liu, B.B.; Li, J.M.; Wang, R.Z.; Zhao, K.J.; Wu, J. Rearrangement and domestication as drivers of Rosaceae mitogenome plasticity. BMC Biol. 2022, 20, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- McCauley, D.E. Paternal leakage, heteroplasmy, and the evolution of plant mitochondrial genomes. New Phytol. 2013, 200, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, W.; Zhang, R.G.; Mao, J.G.; Wang, X.R. Repetitive Elements, Sequence Turnover and Cyto-Nuclear Gene Transfer in Gymnosperm Mitogenomes. Front. Genet. 2022, 13, 867736. [Google Scholar] [CrossRef]
- Bi, C.; Paterson, A.H.; Wang, X.L.; Xu, Y.Q.; Wu, D.Y.; Qu, Y.S.; Jiang, A.N.; Ye, Q.L.; Ye, N. Analysis of the Complete Mitochondrial Genome Sequence of the Diploid Cotton Gossypium raimondii by Comparative Genomics Approaches. Biomed Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef]
- D’Errico, I.; Gadaleta, G.; Saccone, C. Pseudogenes in metazoa: Origin and features. Brief. Funct. Genom. Proteomic. 2004, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.; Priyadarshani, S.; Wang, Y.; Qin, Y. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda Glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.; Sloan, D. Plant Mitochondrial Genome Diversity: The Genomics Revolution; Springer: Dordrecht, Germany, 2012; Volume 9, pp. 123–144. [Google Scholar]
- Dong, S.S.; Zhao, C.X.; Chen, F.; Liu, Y.H.; Zhang, S.Z.; Wu, H.; Zhang, L.S.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Ma, Y.; Kou, L.; Wei, J.; Wang, W. The complete mitochondrial genome of okra (Abelmoschus esculentus): Using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genom. 2022, 23, 481. [Google Scholar] [CrossRef]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cuthbert, J.M.; Taylor, D.R.; Sloan, D.B. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef]
- Guo, W.; Grewe, F.; Fan, W.S.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes Reveal Extreme Contrasts in Gymnosperm Mitochondrial Evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef]
- Fang, B.; Li, J.; Zhao, Q.; Liang, Y.; Yu, J. Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes 2021, 12, 1970. [Google Scholar] [CrossRef]
- Jaime, I.; Arrieta-Montiel, M.P.; Wamboldt, Y.; Cao, J.; Hagmann, J.; Shedge, V.; Xu, Y.-Z.; Weigel, D.; A Mackenzie, S. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 2011, 9, 14. [Google Scholar]
- Marie Miller-Messmer, K.K.P.I.; Jose, M. Gualberto. RecA-Dependent DNA Repair Results in Increased Heteroplasmy of the Arabidopsis Mitochondrial Genome. Plant Physiol. 2012, 159, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Woloszynska, M.; Trojanowski, D. Counting mtDNA molecules in Phaseolus vulgaris: Sublimons are constantly produced by recombination via short repeats and undergo rigorous selection during substoichiometric shifting. Plant Mol. Biol. 2009, 70, 511–521. [Google Scholar] [CrossRef]
- Mizuho Satoh, T.K.; Mikami, T. The Owen mitochondrial genome in sugar beet (Beta vulgaris L.): Possible mechanisms of extensive rearrangements and the origin of the mitotype-unique regions. Appl. Genet. 2006, 113, 8. [Google Scholar]
- Wallet, C.; Le Ret, M.; Bergdoll, M.; Bichara, M.; Dietrich, A.; Gualberto, J.M. The RECG1 DNA Translocase Is a Key Factor in Recombination Surveillance, Repair, and Segregation of the Mitochondrial DNA in Arabidopsis. Plant Cell 2015, 27, 2907–2925. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C.; et al. Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genom. 2005, 272, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Hazle, T.; Bonen, L. Status of genes encoding the mitochondrial S1 ribosomal protein in closely-related legumes. Gene 2007, 405, 108–116. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Accession Number | Contigs | Type | Length | GC Content |
|---|---|---|---|---|
| OL450370-OL450371 | Chromosome 1-2 | Branched | 436,464 bp | 45.20% |
| OL450370 | Chromosome 1 | Circular | 307,720 bp | 45.15% |
| OL450371 | Chromosome 2 | Circular | 128,744 bp | 45.33% |
| Group of Genes | Name of Genes |
|---|---|
| ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| NADH dehydrogenase | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 |
| Cytochrome c biogenesis | cob |
| Ubiquinol cytochrome c reductase | ccmB, ccmC, ccmFC, ccmFN |
| Cytochrome c oxidase | cox1, cox2, cox3 |
| Maturases | matR |
| Transport membrane protein | mttB |
| Large subunit of ribosome | rpl5, rpl10, rpl16 |
| Small subunit of ribosome | rps1, rps3, rps4, rps12, rps13 |
| Succinate dehydrogenase | sdh4 |
| Ribosome RNA | rrn5, rrn18, rrn26 |
| Transfer RNA | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA (×3), trnG-GCC, trnH-GUG, trnK-UUU, trnL-CAA, trnM-CAU (×3), trnN-GUU, trnP-UGG, trnQ-UUG, trnS-GGA, trnS-UGA, trnT-GUA, trnV-GAC, trnW-CCA, trnY-GUA |
| ID | Length (bp) | Identity (%) | Position | Number of Supported Reads | Percentage (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Major 1 | Major 2 | Alternative 1 | Alternative 2 | Major Conformation | Alternative ConformatioN | ||||
| R1 | 7887 | 100 | chr1: 299834-307720; chr1: 107714-115600 | 16 | 18 | 21 | 20 | 0.4533 | 0.5467 |
| R2 | 1235 | 100 | chr1: 63771-65005; chr2: 1-1235 | 168 | 100 | 3 | 0 | 0.9889 | 0.0111 |
| R4 | 331 | 100 | chr1: 48854-48524; chr1: 116771-117101 | 161 | 188 | 1 | 0 | 0.9971 | 0.0029 |
| R5 | 244 | 100 | chr1: 12185-12428; chr1: 33887-34130 | 244 | 206 | 0 | 1 | 0.9978 | 0.0022 |
| R7 | 163 | 100 | chr1: 107876-107714; chr2: 121991-122153 | 242 | 237 | 2 | 0 | 0.9958 | 0.0042 |
| R14 | 163 | 100 | chr1: 299996-299834; chr2: 121991-122153 | 17 | 26 | 1 | 0 | 0.9773 | 0.0227 |
| R15 | 89 | 96.629 | chr1: 169238-169150; chr1: 207012-207100 | 228 | 146 | 1 | 0 | 0.9973 | 0.0027 |
| Chromosome | Order in Contigs |
|---|---|
| Contig 1 | 3-R12-1-R12-12-R10-10-R11-8-R4-6 |
| Contig 2 | 27-R13-31-R7/R1-R7-26-R8-25-R11-24-R9-23-R6-30-R3-29-R6-28 |
| Contig 3 | 22-R3-21-R5-20-R5-19-R9-18-R4-17-R8-16-R13-15 |
| Contig 4 | 14-R10-13-R7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, P.; Huang, Y.; Zong, M.; Xu, Z. De Novo Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Chaenomeles speciosa (Sweet) Nakai Revealed the Existence of Two Structural Isomers. Genes 2023, 14, 526. https://doi.org/10.3390/genes14020526
Cao P, Huang Y, Zong M, Xu Z. De Novo Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Chaenomeles speciosa (Sweet) Nakai Revealed the Existence of Two Structural Isomers. Genes. 2023; 14(2):526. https://doi.org/10.3390/genes14020526
Chicago/Turabian StyleCao, Pei, Yuan Huang, Mei Zong, and Zilong Xu. 2023. "De Novo Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Chaenomeles speciosa (Sweet) Nakai Revealed the Existence of Two Structural Isomers" Genes 14, no. 2: 526. https://doi.org/10.3390/genes14020526
APA StyleCao, P., Huang, Y., Zong, M., & Xu, Z. (2023). De Novo Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Chaenomeles speciosa (Sweet) Nakai Revealed the Existence of Two Structural Isomers. Genes, 14(2), 526. https://doi.org/10.3390/genes14020526