The Impact of Variation in the Toll-like Receptor 3 Gene on Epizootic Hemorrhagic Disease in Illinois Wild White-Tailed Deer (Odocoileus virginianus)

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
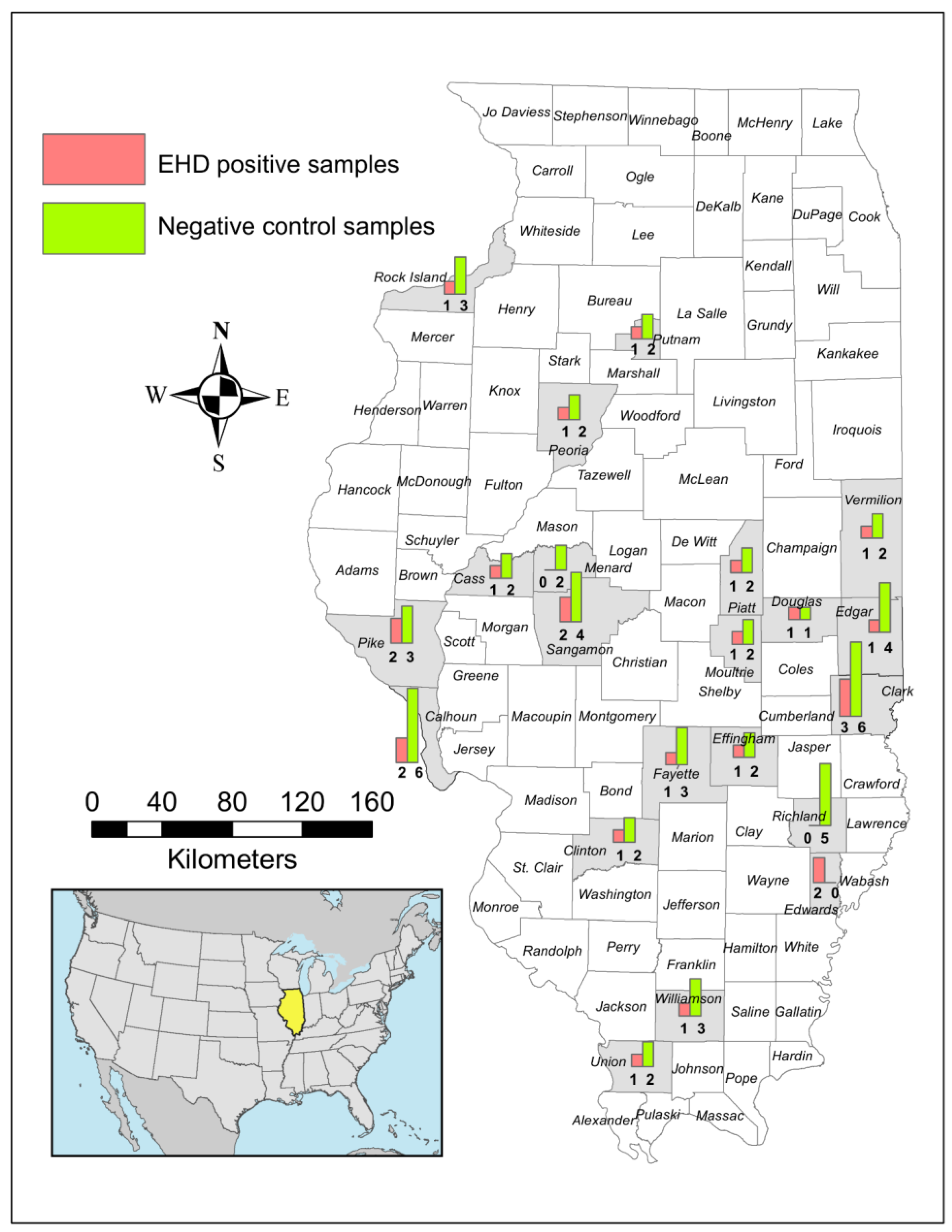
2.1. Study Area and Sample Collection
2.2. Exon Determination and Primer Design
2.3. DNA Extraction, PCR, and Sanger Sequencing of the TLR3 Gene
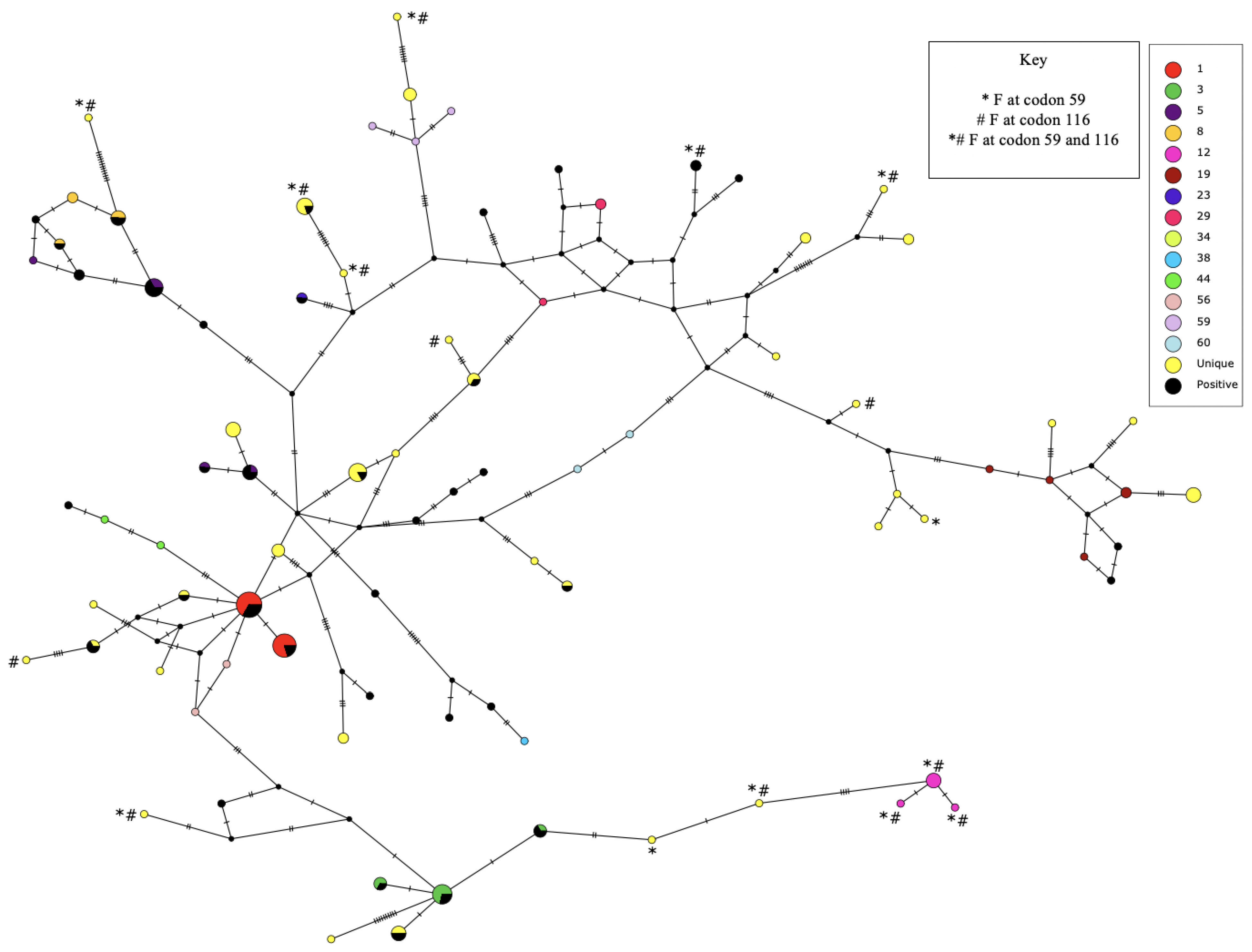
2.4. Single Nucleotide Polymorphism (SNP) Analyses
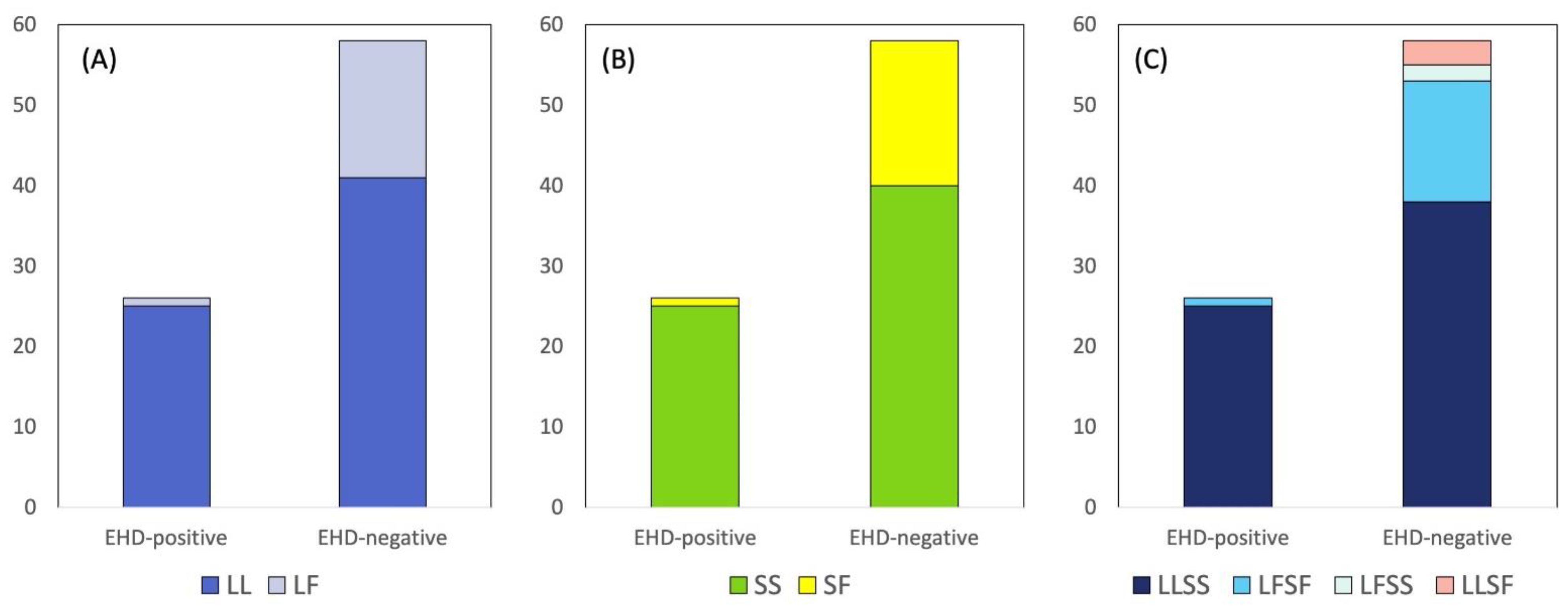
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mcvey, S.D.; Maclachlan, J.N. Vaccines for prevention of bluetongue and epizootic hemorrhagic disease in livestock: A North American perspective. Vector Borne Zoonotic Dis. 2015, 15, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Ruder, M.G.; Lysyk, T.J.; Stallknecht, D.E.; Foil, L.D.; Johnson, D.J.; Chase, C.C.; Dargatz, D.A.; Gibbs, E.P.J. Transmission and epidemiology of bluetongue and epizootic hemorrhagic disease in North America: Current perspectives, research gaps, and future directions. Vector Borne Zoonotic Dis. 2015, 15, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Savini, G.; Afonso, A.; Mellor, P.; Aradaib, I.; Yadin, H.; Sanaa, M.; Wilson, W.; Monaco, F.; Domingo, M. Epizootic heamorragic disease. Res. Vet. Sci. 2011, 91, 1–17. [Google Scholar] [CrossRef]
- Kameke, D.; Kampen, H.; Walther, D. Activity of Culicoides spp. (Diptera: Ceratopogonidae) inside and outside of livestock stables in late winter and spring. Parasitol. Res. 2017, 116, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Dorak, S.J.; Varga, C.; Ruder, M.G.; Gronemeyer, P.; Rivera, N.A.; Dufford, D.R.; Skinner, D.J.; Roca, A.L.; Novakofski, J.; Mateus-Pinilla, N.E. Spatial epidemiology of hemorrhagic disease in Illinois wild white-tailed deer. Sci. Rep. 2022, 12, 6888. [Google Scholar] [CrossRef] [PubMed]
- Brown-Joseph, T.; Rajko-Nenow, P.; Hicks, H.; Sahadeo, N.; Harrup, L.E.; Carrington, C.V.; Batten, C.; Oura, C.A.L. Identification and characterization of epizootic hemorrhagic disease virus serotype 6 in cattle co-infected with bluetongue virus in Trinidad, West Indies. Vet. Microbiol. 2019, 229, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maclachlan, N.J.; Zientara, A.; Savini, G.; Daniels, P.W. Epizootic haemorrhagic disease. Rev. Sci. Tech. Off. Int. Epiz. 2015, 34, 341–351. [Google Scholar] [CrossRef]
- Allison, A.B.; Goekjian, V.H.; Potgieter, A.C.; Wilson, W.C.; Johnson, D.J.; Mertens, P.P.C.; Stallknecht, D.E. Detection of a novel reassortant epizootic hemorrhagic disease virus (EHDV) in the USA containing RNA segments derived from both exotic (EHDV-6) and endemic (EHDV-2) serotypes. J. Gen. Virol. 2010, 91, 430–439. [Google Scholar] [CrossRef]
- Rivera, N.A.; Varga, C.; Ruder, M.G.; Dorak, S.J.; Roca, A.L.; Novakofski, J.E.; Mateus-Pinilla, N.E. Bluetongue and Epizootic Hemorrhagic Disease in the United States of America at the Wildlife–Livestock Interface. Pathogens 2021, 10, 915. [Google Scholar] [CrossRef]
- Howerth, E.W.; Stallknecht, D.E.; Kirkland, P.D. Bluetongue, epizootic haemorrhagic disease, and other orbivirus-related diseases. In Infectious Diseases of Wild Mammals; Williams, E.S., Barker, I.K., Eds.; Iowa State University Press: Ames, IA, USA, 2001; pp. 77–97. [Google Scholar]
- Pfeifer, M. Epizootic Hemorrhagic Disease (EHD) in White-Tailed Deer. Texas A&M Veterinary Medical Diagnostic Laboratory. Available online: https://tvmdl.tamu.edu/2019/07/29/epizootic-hemorrhagic-disease-ehd-in-white-tailed-deer (accessed on 25 October 2022).
- Trejo-de la O, A.; Hernández-Sancén, P.; Maldonado-Bernal, C. Relevance of single-nucleotide polymorphisms in human TLR genes to infectious and inflammatory diseases and cancer. Genes Immun. 2014, 15, 199–209. [Google Scholar] [CrossRef]
- Skevaki, C.; Pararas, M.; Kostelidou, K.; Tsakris, A.; Routsias, J.G. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious diseases. Clin. Exp. Immun. 2015, 180, 165–177. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.; Massari, P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef]
- Chuang, T.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta. 2001, 1518, 157–161. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Gosch, S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Yabsley, M.J.; Howerth, E.W. Initial sequencing and tissue distribution of Toll-like receptor 3 mRNA in white-tailed deer (Odocoileus virginianus). J. Wildl. Dis. 2009, 45, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.A.; Bhattarai, E.K.; Osterstock, J.B.; Dowd, S.E.; Seabury, P.M.; Vikram, M.; Whitlock, R.H.; Schukken, Y.H.; Schnabel, R.D.; Taylor, J.F.; et al. Evolution of the bovine TLR gene family and member associations with Mycobacterium avium subspecies paratuberculosis infection. PLoS ONE 2011, 6, e27744. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Expasy Translate. Swiss Institute of Bioinformatics. Available online: https://web.expasy.org/translate/ (accessed on 25 October 2022).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.; Wink, M. Direct DNA sequencing of PCR-amplified vector inserts following enzymatic degradation of primer and dNTPs. BioTechniques 1994, 17, 858. [Google Scholar] [PubMed]
- Stephens, M.; Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J. Sequence Polymorphism Analysis using DnaSP. In Bioinformatics for DNA Sequence Analysis; Methods in Molecular Biology Series; Posada, D., Ed.; Humana Press: Hatfield, UK, 2009; Volume 537, pp. 337–350. [Google Scholar]
- Kumar, S.; Stetcher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bandelt, H.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Brysbaert, M. How many participants do we have to include in properly powered experiments? J. Cogn. 2019, 2, 16. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org/ (accessed on 6 July 2021).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Provean Version 1.1.3, J. Craig Venter Institute. Available online: http://provean.jcvi.org/index.php (accessed on 25 October 2022).
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Bell, J.K.; Botos, I.; Hall, P.R.; Askins, J.; Shiloach, J.; Segal, D.M.; Davies, D.R. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc. Natl. Acad. Sci. USA 2005, 102, 10976–10980. [Google Scholar] [CrossRef]
- Choe, J.; Kelker, M.S.; Wilson, I.A. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 2005, 309, 581–585. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Taylor, M.; Hammers, M.; Burke, T.; Komdeur, J.; Dugdale, H.; Richardson, D. Contemporary evolution of the viral-sensing TLR3 gene in an isolated vertebrate population. Authorea Prepr. 2021. [Google Scholar] [CrossRef]
- Knafler, G.J.; Ortiz-Catedral, L.; Jackson, B.; Varsani, A.; Grueber, C.E.; Robertson, B.C.; Jamieson, I.G. Comparison of beak and feather disease virus prevalence and immunity-associated genetic diversity over time in an island population of red-crowned parakeets. Arch. Virol. 2016, 161, 811–820. [Google Scholar] [CrossRef]
- Abrantes, J.; Areal, H.; Esteves, P.J. Insights into the European rabbit (Oryctolagus cuniculus) innate immune system: Genetic diversity of the toll-like receptor 3 (TLR3) in wild populations and domestic breeds. BMC Genet. 2013, 14, 73. [Google Scholar] [CrossRef]
- Quéméré, E.; Galan, M.; Cosson, J.F.; Klein, F.; Aulagnier, S.; Gilot-Fromont, E.; Merlet, J.; Bonhomme, M.; Hewison, A.M.; Charbonnel, N. Immunogenetic heterogeneity in a widespread ungulate: The European roe deer (Capreolus capreolus). Mol. Ecol. 2015, 24, 3873–3887. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Y.; Li, C.; Li, R.; Xiao, H.; Chen, S. Genetic diversity of TLR3 and TLR8 genes among five Chinese native cattle breeds from Southwest China. Livest. Sci. 2020, 232, 103895. [Google Scholar] [CrossRef]
- Gaydos, J.K.; Davidson, W.R.; Elvinger, F.; Howerth, E.W.; Murphy, M.; Stallknecht, D.E. Cross-protection between epizootic hemorrhagic disease virus serotypes 1 and 2 in white-tailed deer. J. Wildl. Dis. 2002, 38, 720–728. [Google Scholar] [CrossRef]
- O’Hara Ruiz, M.; Kelly, A.C.; Brown, M.W.; Novakofski, J.E.; Mateus-Pinilla, N.E. Influence of landscape factors and management decisions on spatial and temporal patterns of the transmission of chronic wasting disease in white-tailed deer. Geospat. Health 2013, 8, 215–227. [Google Scholar] [CrossRef]
- Kelly, A.C.; Mateus-Pinilla, N.E.; Brown, W.; Ruiz, M.O.; Douglas, M.R.; Douglas, M.E.; Shelton, P.; Beissel, T.; Novakofski, J. Genetic assessment of environmental features that influence deer dispersal: Implications for prion-infected populations. Popul. Ecol. 2014, 56, 327–340. [Google Scholar] [CrossRef]
- Candela, M.G.; Serrano, E.; Sevila, J.; León, L.; Caro, M.R.; Verheyden, H. Pathogens of zoonotic and biological importance in roe deer (Capreolus capreolus): Seroprevalence in an agro-system population in France. Res. Vet. Sci. 2014, 96, 254–259. [Google Scholar] [CrossRef]
- Quéméré, E.; Hessenauer, P.; Galan, M.; Fernandez, M.; Merlet, J.; Chaval, Y.; Morellet, N.; Verheyden, H.; Gilot-Fromont, E.; Charbonnel, N. Pathogen-mediated selection favours the maintenance of innate immunity gene polymorphism in a widespread wild ungulate. J. Evol. Biol. 2021, 34, 1156–1166. [Google Scholar] [CrossRef]
- Yang, X.; Joyee, A. Role of toll-like receptors in immune responses to chlamydial infections. Curr. Pharm. Des. 2008, 14, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Mun, H.-S.; Aosai, F.; Norose, K.; Chen, M.; Piao, L.-X.; Takeuchi, O.; Akira, S.; Ishikura, H.; Yano, A. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int. Immunol. 2003, 15, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Heni, A.C.; Schmid, J.; Rasche, A.; Corman, V.M.; Drosten, C.; Sommer, S. Pathogen-associated selection on innate immunity genes (TLR4, TLR7) in a neotropical rodent in landscapes differing in anthropogenic disturbance. Heredity 2020, 125, 184–199. [Google Scholar] [CrossRef]
- Kloch, A.; Wenzel, M.A.; Laetsch, D.R.; Michalski, O.; Bajer, A.; Behnke, J.M.; Welc-Falęciak, R.; Piertney, S.B. Signatures of balancing selection in toll-like receptor (TLRs) genes–novel insights from a free-living rodent. Sci. Rep. 2018, 8, 8361. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Exon Targeted | Primer Name | Primer Sequence (5’ to 3’) | Uses | |
|---|---|---|---|---|
| Exon 2 | OdVi-TLR3-X2 | F1: | GGGAAGGGGGAGAGTTTGTA | PCR and Sequencing |
| R1: | CATATTTGAGTGTGGGGTCCC | PCR and Sequencing | ||
| Exon 3 | OdVi-TLR3-X3 | F3: | CCATTTTGGTGCCAAGACTA | PCR and Sequencing |
| R4: | AGCCCAGACAGGAAATCAGC | PCR and Sequencing | ||
| Exon 4 | OdVi-TLR3-X4 | F1: | GATCAGGGAAGACCCTCTGA | PCR and Sequencing |
| R1: | CCAGAGCCGAGCTAAGTTGT | PCR and Sequencing | ||
| SF1: | CAAGCTGAGCCCCAGTCTC | Sequencing Only | ||
| SR1: | GTCTGCTTCAGTCCATCGAA | Sequencing Only | ||
| SF2: | CGCTCTTTTTATGGGCTTTC | Sequencing Only | ||
| SR2: | GCCACTGAAAGGAAAAATCG | Sequencing Only | ||
| SF3: | GCCACTGAAAGGAAAAATCG | Sequencing Only | ||
| SR3: | TCATTTGTTAAAGTCCGCAAA | Sequencing Only | ||
| SF4: | AGCTGACCACCAACTCTTTCA | Sequencing Only | ||
| SR4 | TTGCTTAGATCCAGAATGACCA | Sequencing Only | ||
| F2: | CCTGGTCATTCTGGATCTAAGC | PCR and Sequencing | ||
| R2: | ATTTCAAATGTCATAGTGTTCACC | PCR and Sequencing | ||
| SF5: | TTTGATGAGATCCCAGTGGA | Sequencing Only | ||
| SR5: | ACAAACCAGGCAATGCTTTC | Sequencing Only | ||
| SF6: | CTGCTCATCCATTTTGAAGG | Sequencing Only | ||
| SR6: | TGCTGCATATTCAAACTGCTC | Sequencing Only | ||
| SF7: | CAGCATCAGAAGGAGCAGAA | Sequencing Only | ||
| SR7: | ATTTCAAATGTCATAGTGTTCACC | Sequencing Only | ||
| Exon 5 | OdVi-TLR3_X5 | F1: | GATTTTAGAGTGTTGGGCTGTT | PCR and Sequencing |
| R1: | AAGGCCTGAAATAGGGAGACA | PCR and Sequencing | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wessels, J.E.; Ishida, Y.; Rivera, N.A.; Stirewalt, S.L.; Brown, W.M.; Novakofski, J.E.; Roca, A.L.; Mateus-Pinilla, N.E. The Impact of Variation in the Toll-like Receptor 3 Gene on Epizootic Hemorrhagic Disease in Illinois Wild White-Tailed Deer (Odocoileus virginianus). Genes 2023, 14, 426. https://doi.org/10.3390/genes14020426
Wessels JE, Ishida Y, Rivera NA, Stirewalt SL, Brown WM, Novakofski JE, Roca AL, Mateus-Pinilla NE. The Impact of Variation in the Toll-like Receptor 3 Gene on Epizootic Hemorrhagic Disease in Illinois Wild White-Tailed Deer (Odocoileus virginianus). Genes. 2023; 14(2):426. https://doi.org/10.3390/genes14020426
Chicago/Turabian StyleWessels, Jacob E., Yasuko Ishida, Nelda A. Rivera, Spencer L. Stirewalt, William M. Brown, Jan E. Novakofski, Alfred L. Roca, and Nohra E. Mateus-Pinilla. 2023. "The Impact of Variation in the Toll-like Receptor 3 Gene on Epizootic Hemorrhagic Disease in Illinois Wild White-Tailed Deer (Odocoileus virginianus)" Genes 14, no. 2: 426. https://doi.org/10.3390/genes14020426
APA StyleWessels, J. E., Ishida, Y., Rivera, N. A., Stirewalt, S. L., Brown, W. M., Novakofski, J. E., Roca, A. L., & Mateus-Pinilla, N. E. (2023). The Impact of Variation in the Toll-like Receptor 3 Gene on Epizootic Hemorrhagic Disease in Illinois Wild White-Tailed Deer (Odocoileus virginianus). Genes, 14(2), 426. https://doi.org/10.3390/genes14020426