
A Founder Intronic Variant in P3H1 Likely Results in Aberrant Splicing and Protein Truncation in Patients of Karen Descent with Osteogenesis Imperfecta Type VIII
 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
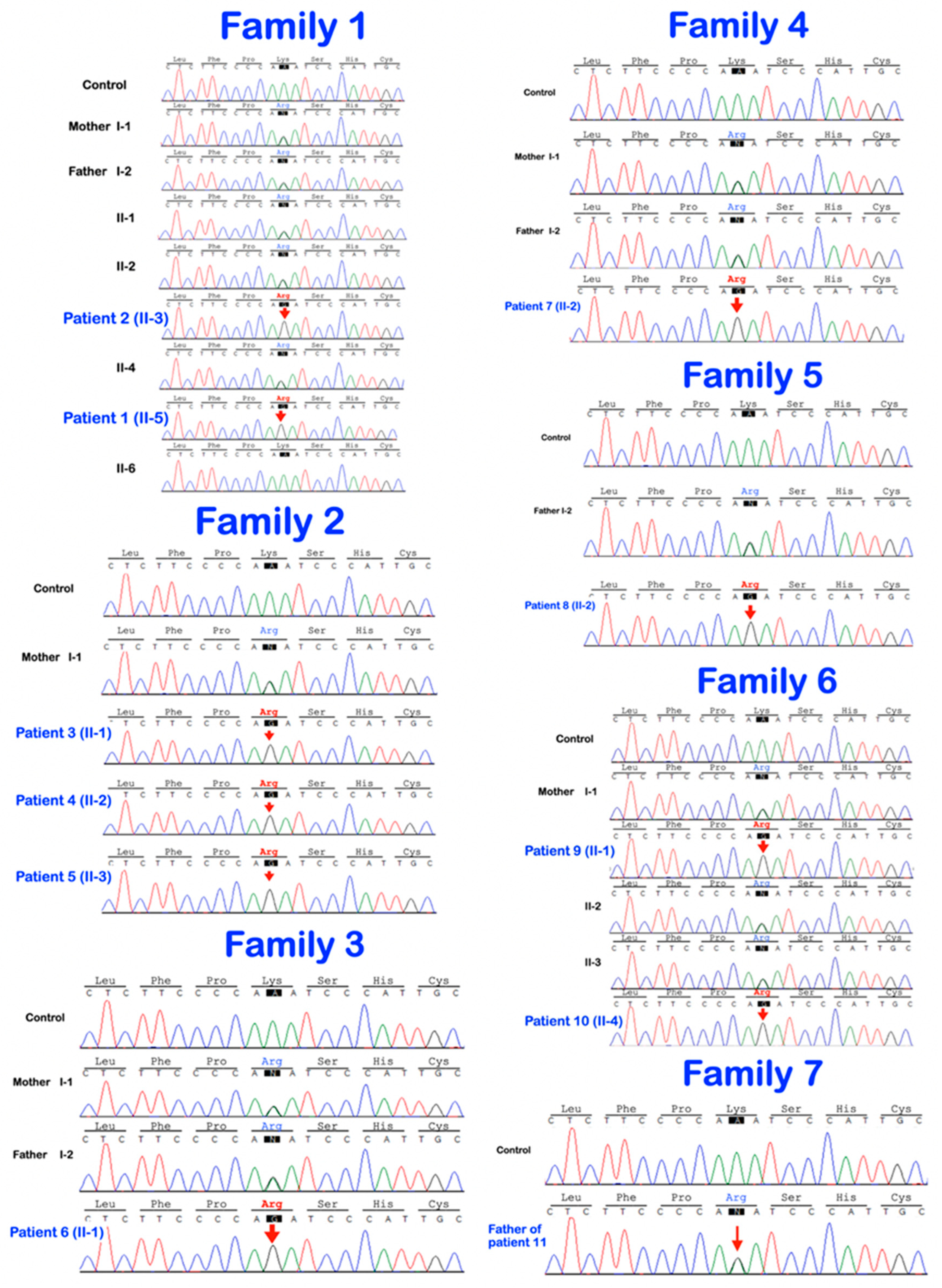
2.2. Whole-Exome Sequencing and Mutation Analysis
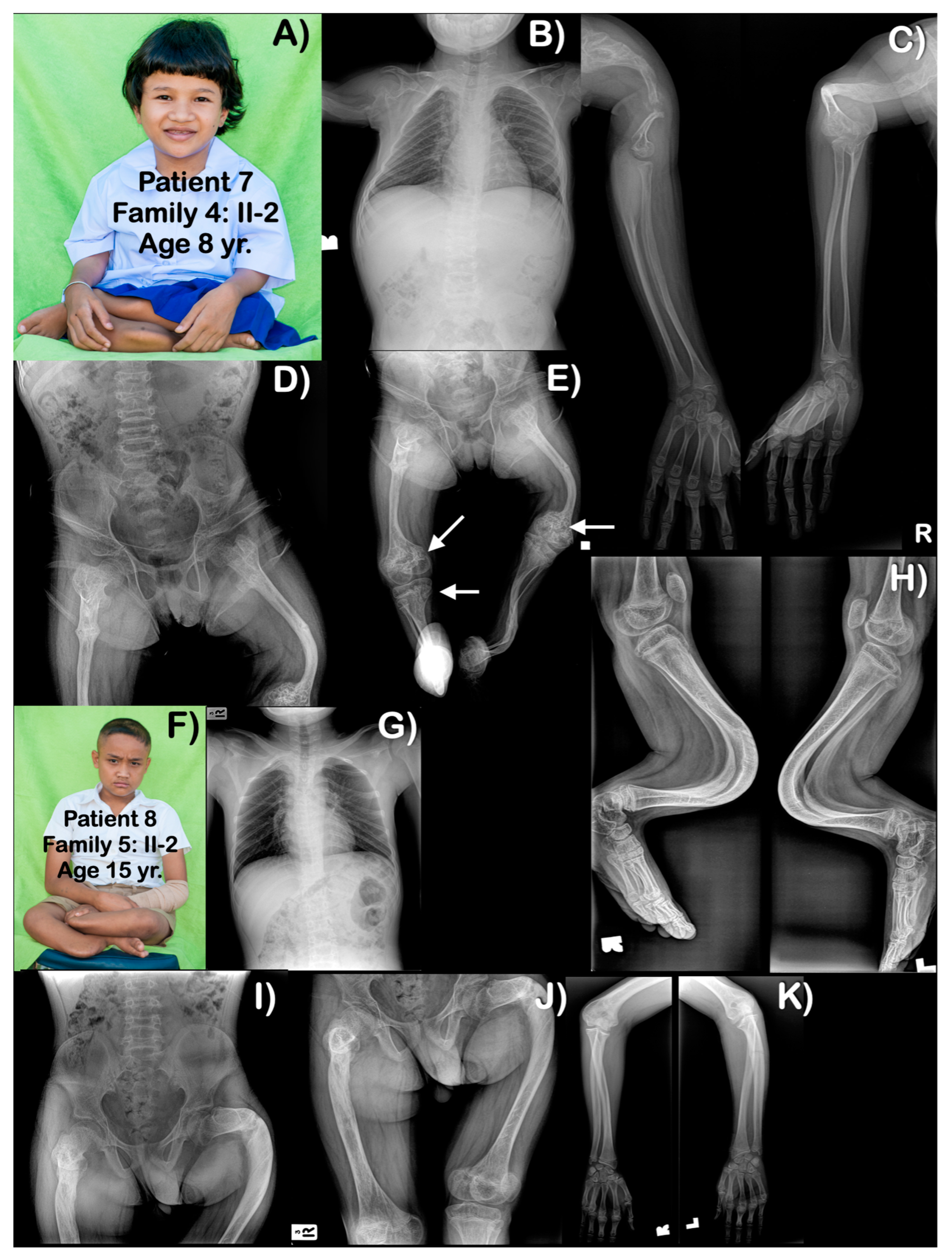
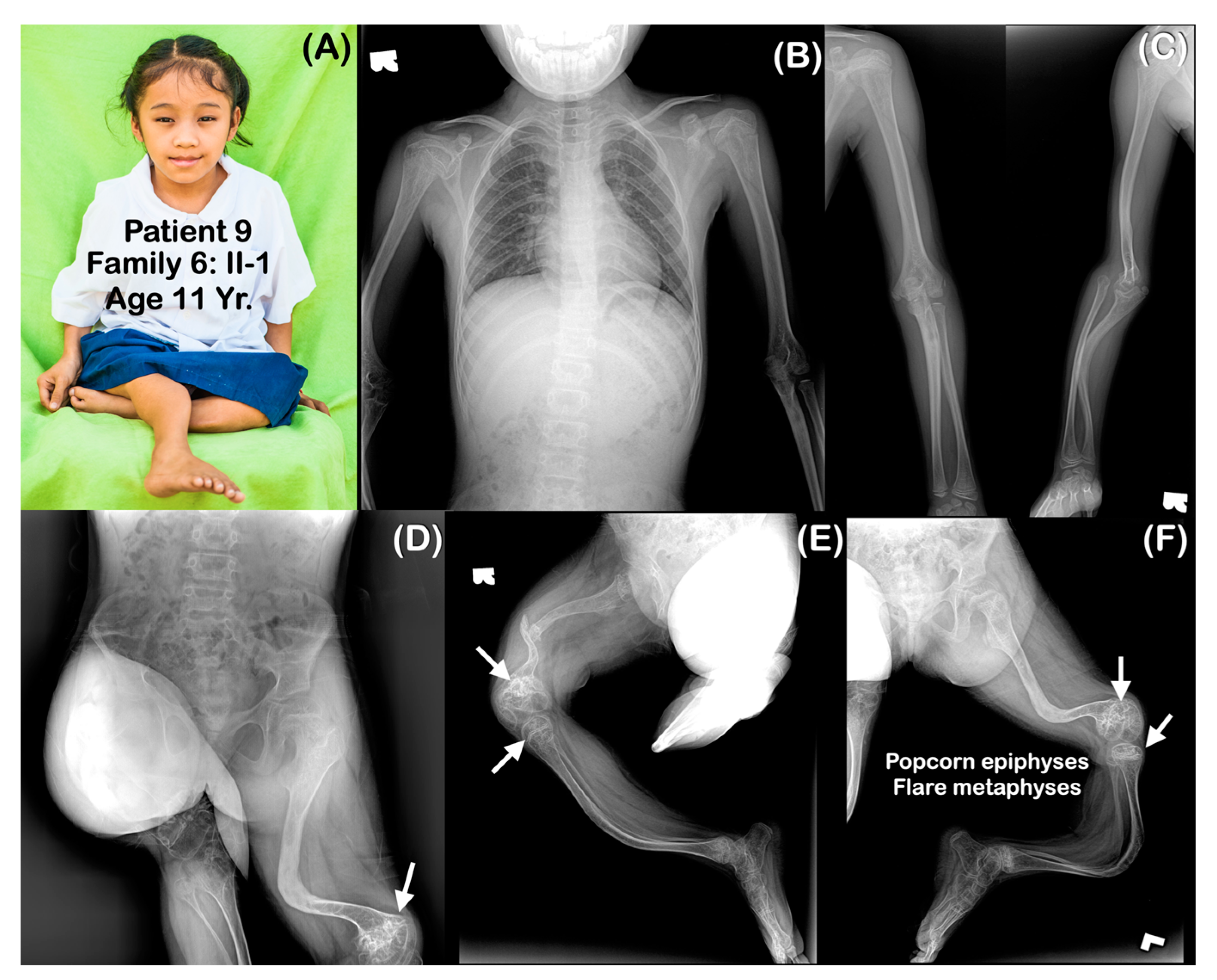
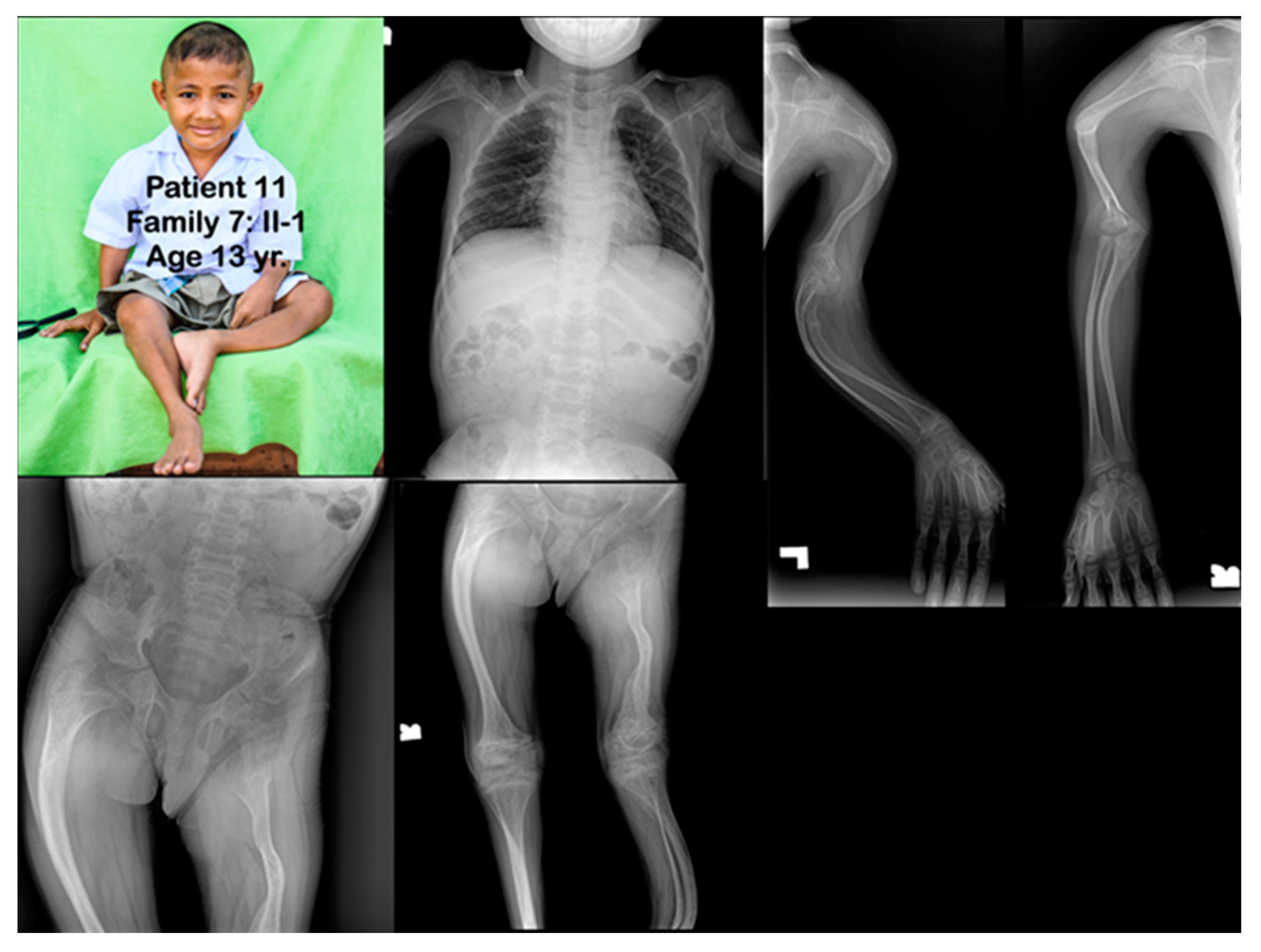
3. Results
4. Discussion
4.1. P3H1 and Its Pathogenic Variants
4.2. Phenotypic Variability
4.3. Popcorn Calcifications
4.4. Rhizomelia
4.5. Bowel Obstruction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marini, J.C.; Cabral, W.A.; Barnes, A.M.; Chang, W. Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 2007, 6, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Wenstrup, R.J.; Willing, M.C.; Starman, B.J.; Byers, P.H. Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am. J. Hum. Genet. 1990, 46, 975–982. [Google Scholar] [PubMed]
- Marini, J.C.; Cabral, W.A.; Barnes, A.M. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010, 339, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Marini, J.C.; Reich, A.; Smith, S.M. Osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr. Opin. Pediatr. 2014, 26, 500–507. [Google Scholar] [CrossRef]
- Takagi, M.; Ishii, T.; Barnes, A.M.; Weis, M.; Amano, N.; Tanaka, M.; Fukuzawa, R.; Nishimura, G.; Eyre, D.R.; Marini, J.C.; et al. A novel mutation in LEPRE1 that eliminates only the KDEL ER- retrieval sequence causes non-lethal osteogenesis imperfecta. PLoS ONE 2012, 7, e36809. [Google Scholar] [CrossRef]
- Cabral, W.A.; Chang, W.; Barnes, A.M.; Weis, M.; Scott, M.A.; Leikin, S.; Makareeva, E.; Kuznetsova, N.V.; Rosenbaum, K.N.; Tifft, C.J.; et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal severe osteogenesis imperfecta. Nat. Genet. 2007, 39, 359–365. [Google Scholar] [CrossRef]
- Vranka, J.A.; Sakai, L.Y.; Bächinger, H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004, 279, 23615–23621. [Google Scholar] [CrossRef]
- Forlino, A.; Cabral, W.A.; Barnes, A.M.; Marini, J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011, 7, 540–557. [Google Scholar] [CrossRef]
- Pokidysheva, E.; Tufa, S.; Bresee, C.; Brigande, J.V.; Bächinger, H.P. Prolyl 3-hydroxylase-1 null mice exhibit hearing impairment and abnormal morphology of the middle ear bone joints. Matrix Biol. 2013, 32, 39–44. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Dejkhamron, P.; Intachai, W.; Ngamphiw, C.; Ketudat Cairns, J.R.; Kawasaki, K.; Ohazama, A.; Olsen, B.; Tongsima, S.; Angkurawaranon, S.A. A novel P3H1 mutation is associated with osteogenesis imperfecta type VIII and dental anomalies. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 132, e198–e207. [Google Scholar] [CrossRef]
- Willaert, A.; Malfait, F.; Symoens, S.; Gevaert, K.; Kayserili, H.; Megarbane, A.; Mortier, G.; Leroy, J.G.; Coucke, P.; De Paepe, A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: Clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009, 46, 233–241. [Google Scholar] [CrossRef]
- Wang, M.; Marin, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef]
- Barnes, A.M.; Chang, W.; Morello, R.; Cabral, W.A.; Weis, M.; Eyre, D.R.; Leikin, S.; Makareeva, E.; Kuznetsova, N.; Uveges, T.E.; et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006, 355, 2757–2764. [Google Scholar] [CrossRef]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef]
- Vranka, J.A.; Pokidysheva, E.; Hayashi, L.; Zientek, K.; Mizuno, K.; Ishikawa, Y.; Maddox, K.; Tufa, S.; Keene, D.R.; Klein, R.; et al. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J. Biol. Chem. 2010, 285, 17253–17262. [Google Scholar] [CrossRef]
- Baldridge, D.; Lennington, J.; Weis, M.; Homan, E.P.; Jiang, M.M.; Munivez, E.; Keene, D.R.; Hogue, W.R.; Pyott, S.; Byers, P.H.; et al. Generalized connective tissue disease in Crtap-/- mouse. PLoS ONE 2010, 5, e10560. [Google Scholar] [CrossRef]
- Baldridge, D.; Schwarze, U.; Morello, R.; Lennington, J.; Bertin, T.K.; Pace, J.M.; Pepin, M.G.; Weis, M.; Eyre, D.R.; Walsh, J.; et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008, 29, 1435–1442. [Google Scholar] [CrossRef]
- Chang, W.; Barnes, A.M.; Cabral, W.A.; Bodurtha, J.N.; Marini, J.C. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum. Mol. Genet. 2010, 19, 223–234. [Google Scholar] [CrossRef]
- Obafemi, A.A.; Bulas, D.I.; Troendle, J.; Marini, J.C. Popcorn calcification in osteogenesis imperfecta: Incidence, progression, and molecular correlation. Am. J. Med. Genet. Part A 2008, 146, 2725–2732. [Google Scholar] [CrossRef]
- Goldman, A.B.; Davidson, D.; Pavlov, H.; Bullough, P.G. “Popcorn” calcifications: A prognostic sign in osteogenesis imperfecta. Radiology 1980, 136, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Stern, T.; Aviram, R.; Rot, C.; Galili, T.; Sharir, A.; Kalish Achrai, N.K.; Ketller, Y.; Shahar, R.; Zelzer, E. Isometric scaling in developing long bones is achieved by an optimal epiphyseal growth balance. PLoS Biol. 2015, 13, e1002212. [Google Scholar] [CrossRef] [PubMed]
- Wenger, D.R.; Abrams, R.A.; Yaru, N.; Leach, J. Obstruction of the colon due to protrusio acetabuli in osteogenesis imperfecta: Treatment by pelvic osteotomy. Report of a case. J. Bone Jt. Surg. Am. 1988, 70, 1103–1107. [Google Scholar] [CrossRef]
- Lee, K.; Park, M.S.; Yoo, W.J.; Chung, C.Y.; Choi, I.H.; Cho, T.J. Proximal migration of femoral telescopic rod in children with osteogenesis imperfecta. J. Pediatr. Orthop. 2015, 35, 178–184. [Google Scholar] [CrossRef]
- Martins, G.; Siedlikowski, M.; Coelho, A.K.S.; Rauch, F.; Tsimicalis, A. Bladder and bowel symptoms experienced by children with osteogenesis imperfecta. J. Pediatr. 2020, 96, 472–478. [Google Scholar] [CrossRef]
- Stockwell, E.; Wallace, M. Obstructive Constipation in Two Patients with Severe Osteogenesis Imperfecta and Acetabular Protrusio. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2022, 6, e21.00194. [Google Scholar] [CrossRef]
- Violas, P.; Fassier, F.; Hamdy, R.; Duhaime, M.; Glorieux, F.H. Acetabular protrusion in osteogenesis imperfecta. J. Pediatr. Orthop. 2002, 22, 622–625. [Google Scholar] [CrossRef]
- Ahn, J.; Carter, E.; Raggio, C.L.; Green, D.W. Acetabular Protrusio in Patients with Osteogenesis Imperfecta: Risk Factors and Progression. J. Pediatr. Orthop. 2019, 39, e750–e754. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
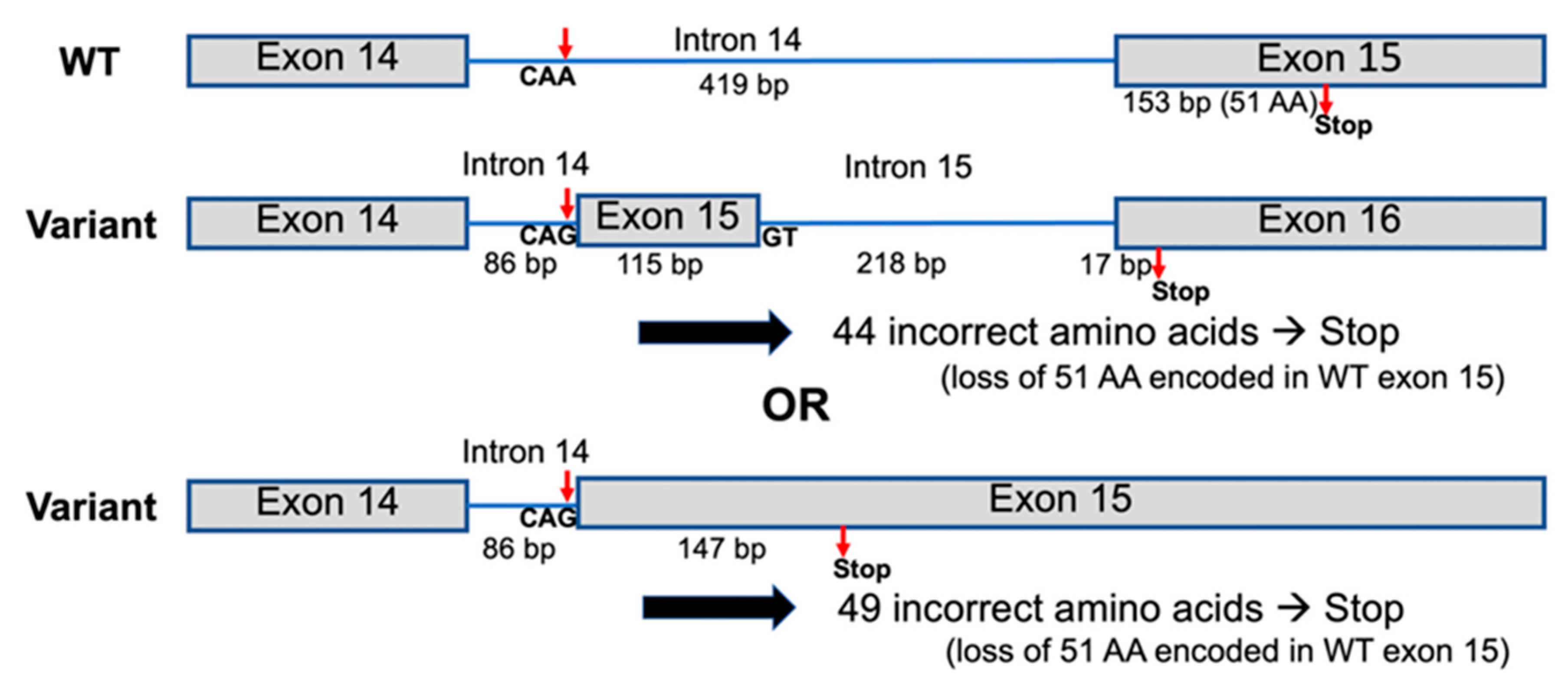{kind=link}
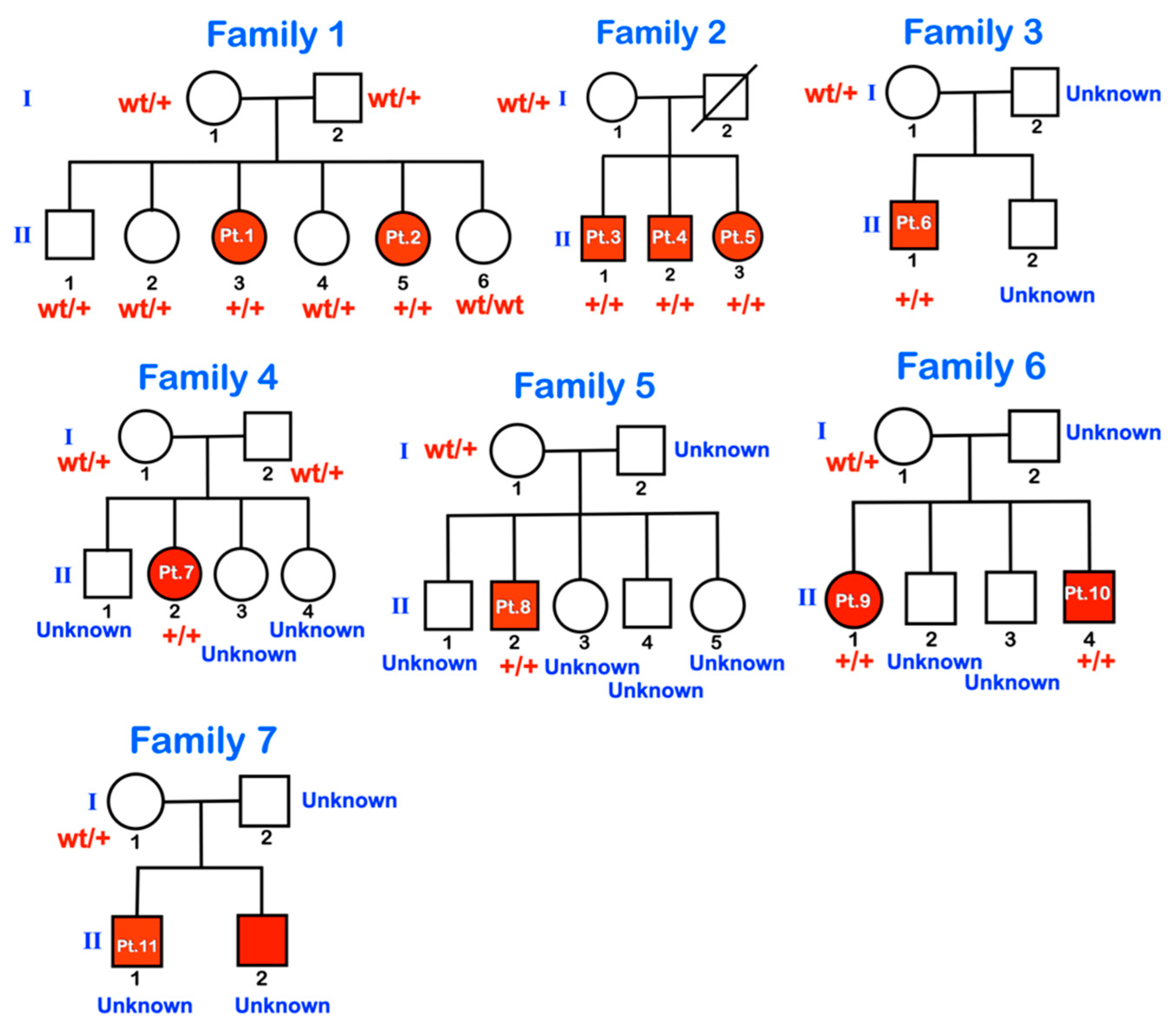
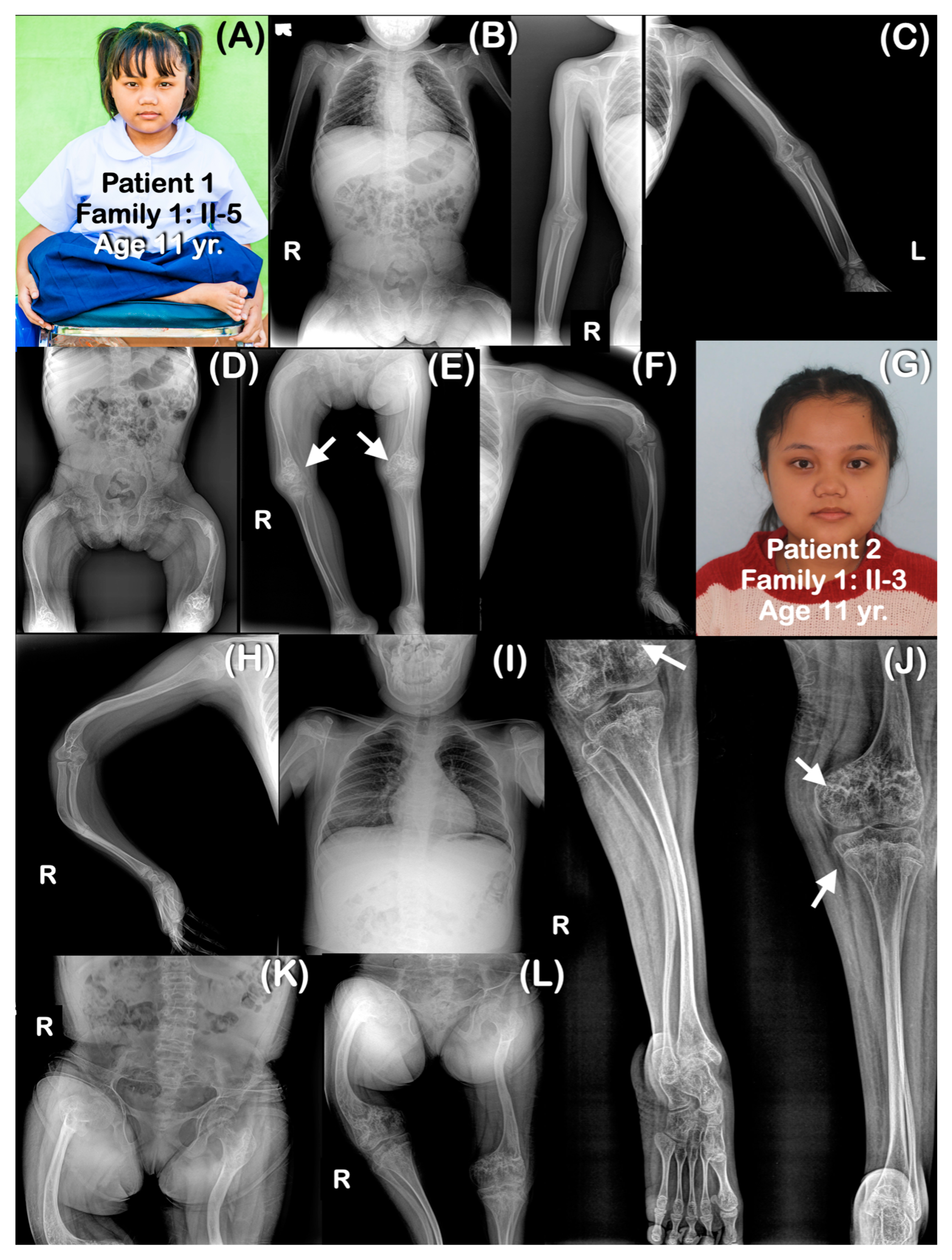
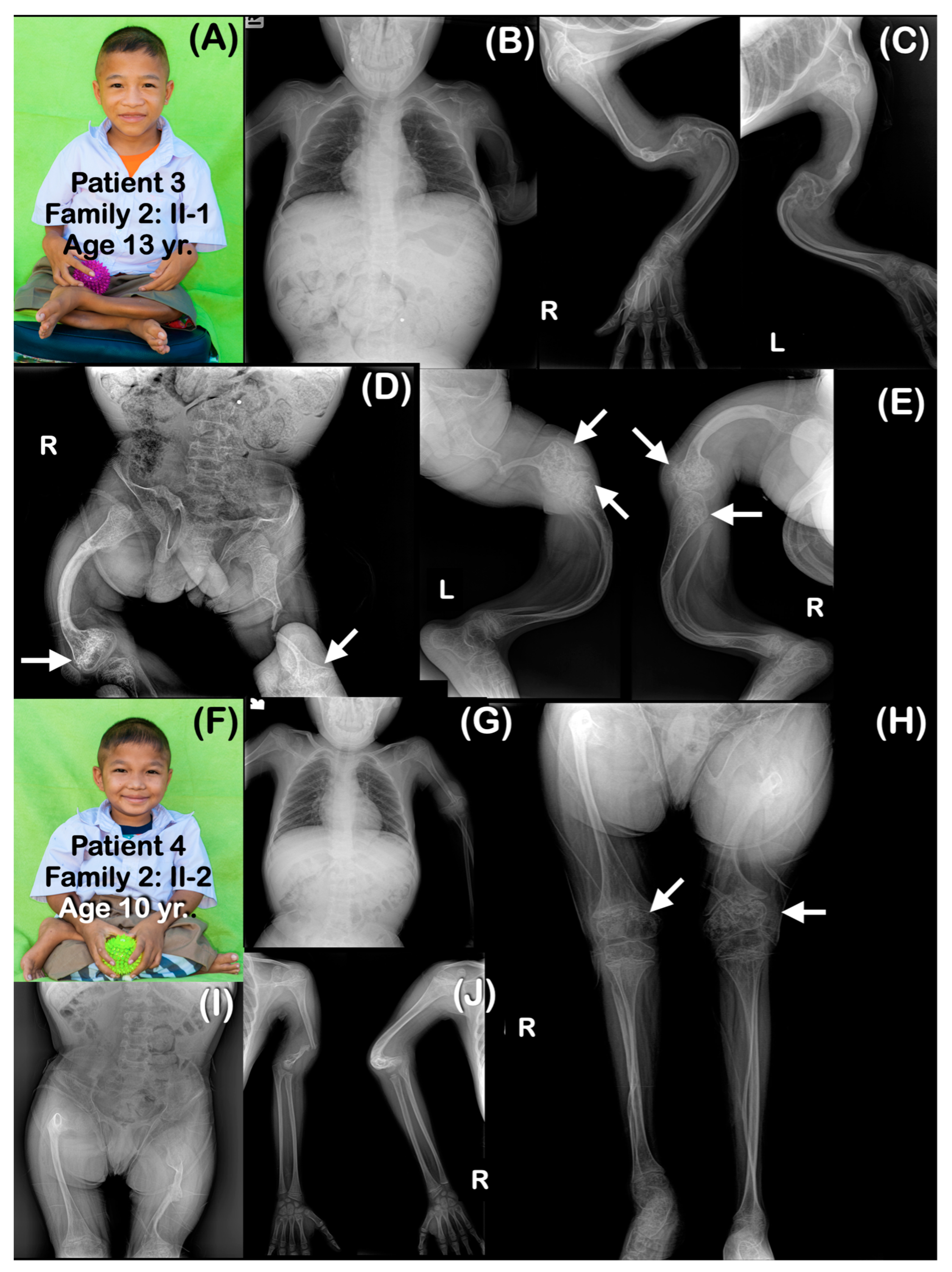
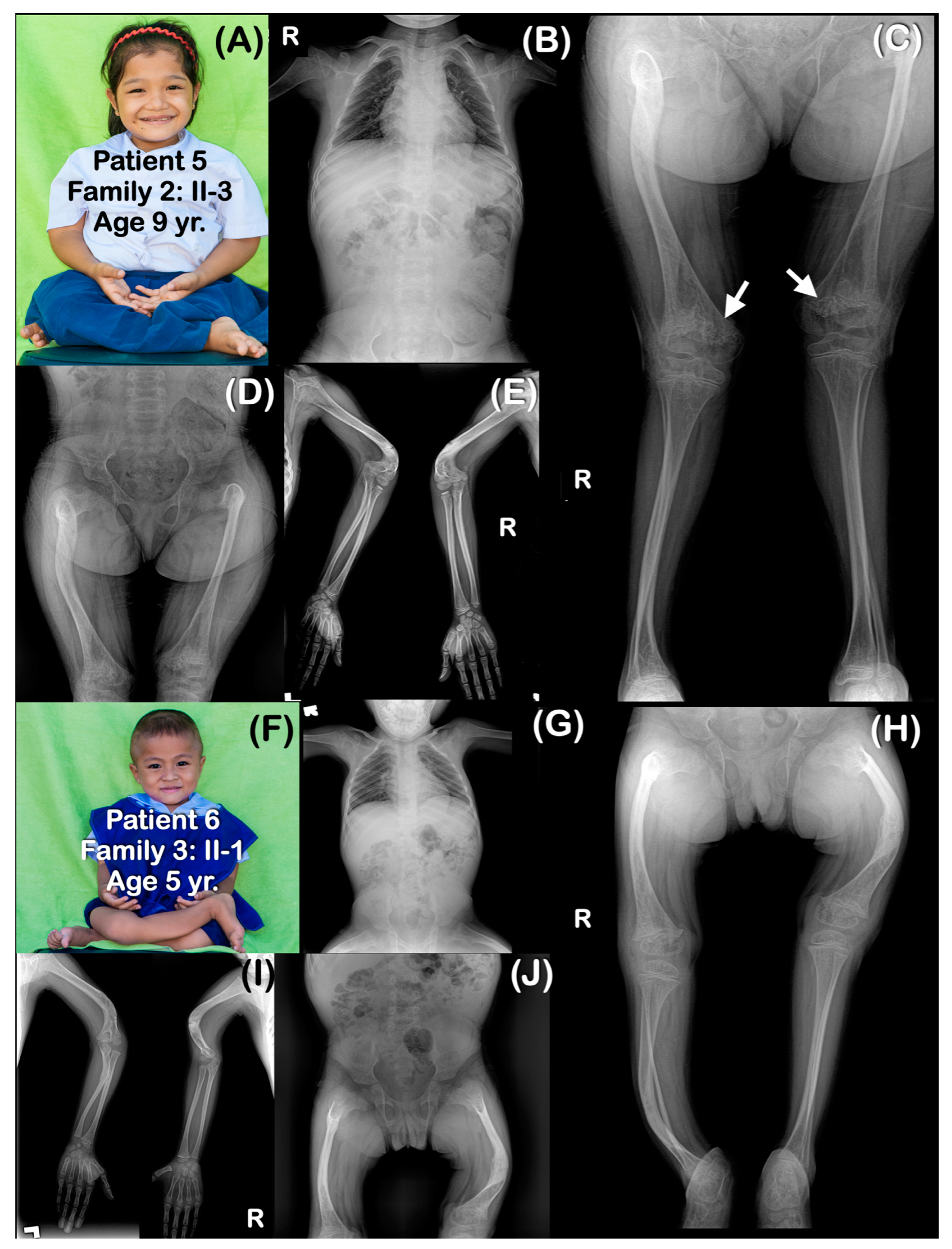
| Families | Pt | Age (yr) | Mutations | Ability to Walk | Generalized Osteopenia | Slender Long Bones | History of Multiple Fractures | Platyspondyly | Blue Slcerae | Rhizomelia | Popcorn Calcification | Remarks |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | 1 | 11 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | No | Yes | |
| 2 | 6 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | No | Yes | ||
| Family 2 | 3 | 13 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | Yes | Yes | Bowel obstruction |
| 4 | 10 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | Yes | Yes | ||
| 5 | 9 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | Yes | Yes | ||
| Family 3 | 6 | 5 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | Yes | No | Absence of some carpal bones |
| Family 4 | 7 | 8 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | Yes | Yes | |
| Family 5 | 8 | 15 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | Yes | No | No | No | |
| Family 6 | 9 | 11 | chr1:43212857A>G (Homozygous) | Never | Yes | Yes | Yes | No | No | No | Yes | |
| 10 | 8 | chr1:43212857A>G (Homozygous) | Never | N/A | N/A | Yes | N/A | N/A | N/A | N/A | ||
| Family 7 | 11 | 13 | N/A His father is heterozygous for the mutation. | Never | Yes | Yes | Yes | Yes | No | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantaputra, P.N.; Angkurawaranon, S.; Intachai, W.; Ngamphiw, C.; Olsen, B.; Tongsima, S.; Cox, T.C.; Ketudat Cairns, J.R. A Founder Intronic Variant in P3H1 Likely Results in Aberrant Splicing and Protein Truncation in Patients of Karen Descent with Osteogenesis Imperfecta Type VIII. Genes 2023, 14, 322. https://doi.org/10.3390/genes14020322
Kantaputra PN, Angkurawaranon S, Intachai W, Ngamphiw C, Olsen B, Tongsima S, Cox TC, Ketudat Cairns JR. A Founder Intronic Variant in P3H1 Likely Results in Aberrant Splicing and Protein Truncation in Patients of Karen Descent with Osteogenesis Imperfecta Type VIII. Genes. 2023; 14(2):322. https://doi.org/10.3390/genes14020322
Chicago/Turabian StyleKantaputra, Piranit Nik, Salita Angkurawaranon, Worrachet Intachai, Chumpol Ngamphiw, Bjorn Olsen, Sissades Tongsima, Timothy C. Cox, and James R. Ketudat Cairns. 2023. "A Founder Intronic Variant in P3H1 Likely Results in Aberrant Splicing and Protein Truncation in Patients of Karen Descent with Osteogenesis Imperfecta Type VIII" Genes 14, no. 2: 322. https://doi.org/10.3390/genes14020322
APA StyleKantaputra, P. N., Angkurawaranon, S., Intachai, W., Ngamphiw, C., Olsen, B., Tongsima, S., Cox, T. C., & Ketudat Cairns, J. R. (2023). A Founder Intronic Variant in P3H1 Likely Results in Aberrant Splicing and Protein Truncation in Patients of Karen Descent with Osteogenesis Imperfecta Type VIII. Genes, 14(2), 322. https://doi.org/10.3390/genes14020322