

Microsatellite Instability and Aberrant Pre-mRNA Splicing: How Intimate Is It?

 ,
,  and
and
Abstract
Foreword
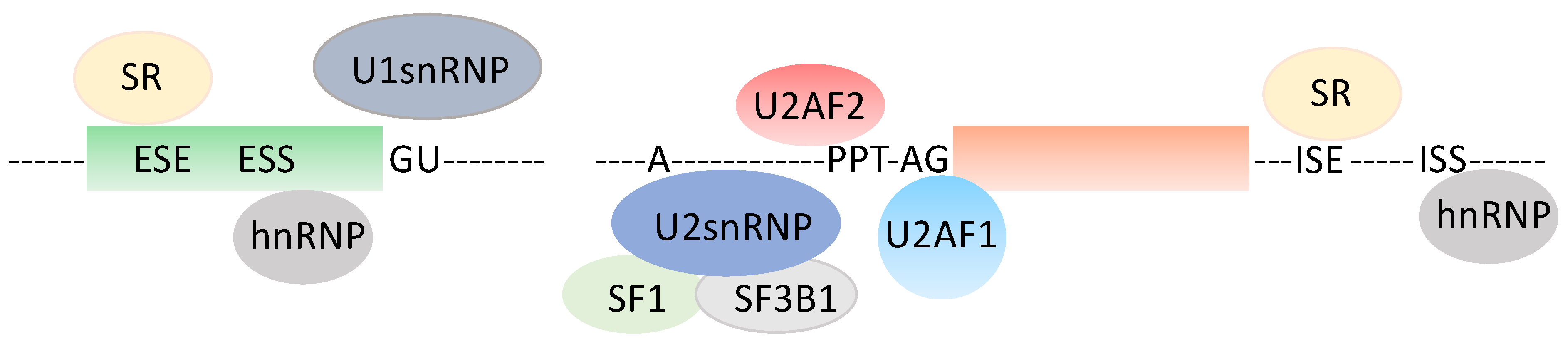
1. A Quick Look at the Pre-mRNA Splicing Unit (Figure 1)

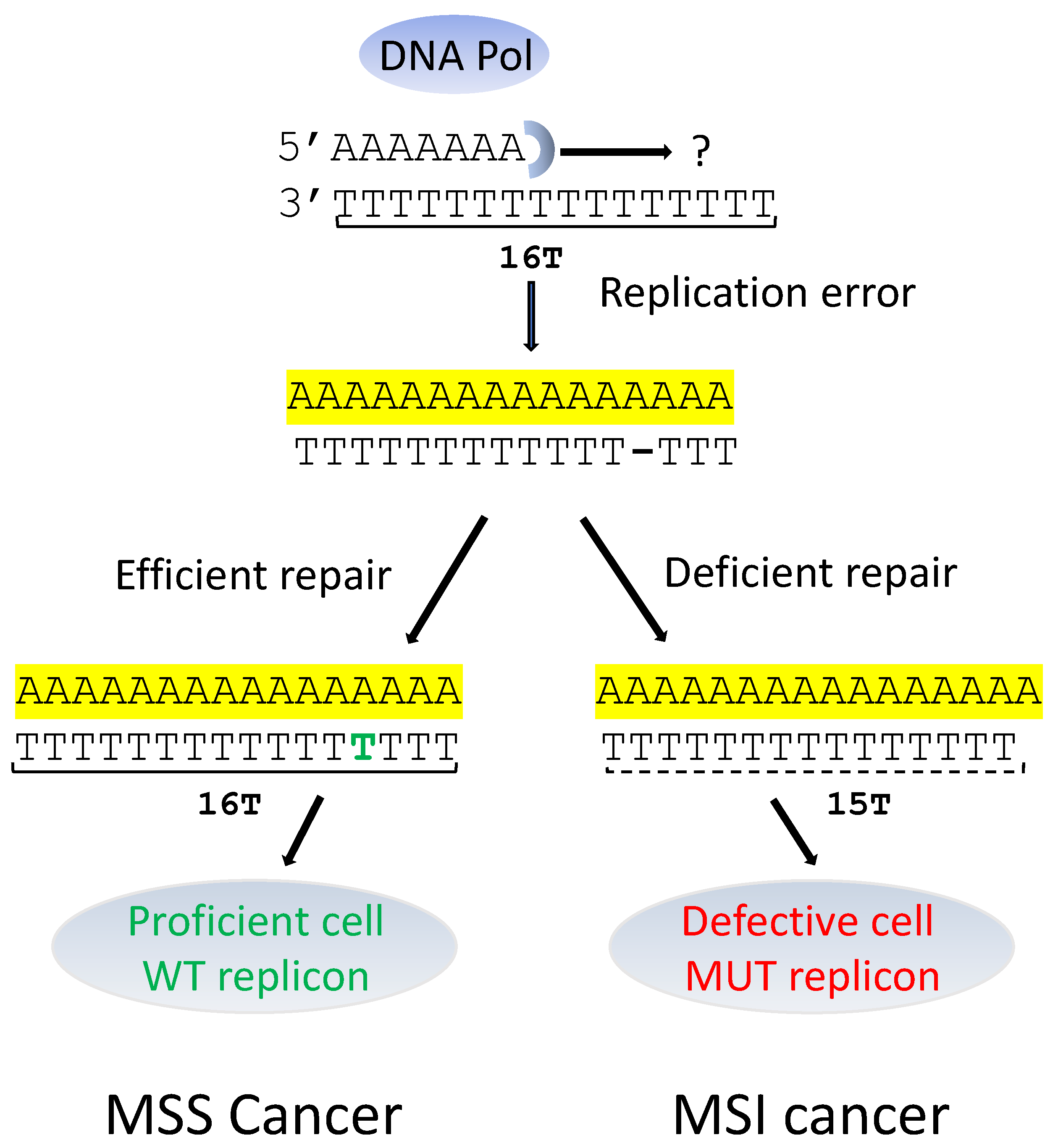
2. Microsatellite Instability Cancers and Pre-mRNA Splicing Alterations (Table 1, Figure 2)

{kind=link}
{kind=link}
{kind=link}
| Gene | Ensembl Transcript | Skipped Exon N° | WT Intronic 3′ End |
|---|---|---|---|
| ATM | ENST00000675843.1 | 6 (GC) | ctaataatttttttttttttttaag |
| ATM | ENST00000675843.1 | 12 (CRC) | tgaagctttttgtttttctttgtag |
| MRE11 | ENST00000323929.8 | 5 | ttttaagtaactttttttttttaag |
| HSP110 | ENST00000320027.10 | 9 | catgatttttttttttttttttaag |

3. Activation of the MMR and DSB Repair Mechanisms (Table 2)
| CDC14 | ADAM28 | PAMR1 | XYLT2 | DNAJC18 | FRMD4 | ATL3 | DDX6 | FHOD3 | ZBTB20 | PSME4 | GSE1 | LARP4B | SCLO6A1 | ZNF43 | PREPL | RNF43 | LMAN1 (e) | LMAN1 (i) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary site | N | MU138468 | MU103116 | MU1871022 | MU62168 | MU158136 | MU61476 | MU71434 | MU69837 | MU59026 | MU92398 | MU56855 | MU81682 | MU89465 | MU73674 | MU57884 | MU93009 | MU70114 | MU5428109 | MU91855 |
| Colon polyps | 66 | 2 | 4 | 4 | 5 | 7 | 7 | 9 | 9 | 11 | 12 | 13 | 14 | 14 | 16 | 17 | 24 | 38 | 40 | 44 |
| Colon | 5 | 3 | 2 | 2 | 1 | 1 | ||||||||||||||
| Stomach | 5 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 3 | 3 | 4 | ||||
| Genital tractus | 14 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 6 | 1 | 2 | 1 | 6 | |||||||
| Others (skin, brain) | 5 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | ||||||||||||
| N = 95 | ||||||||||||||||||||
4. Crossroads between DNA Repair and Pre-mRNA Splicing Programs
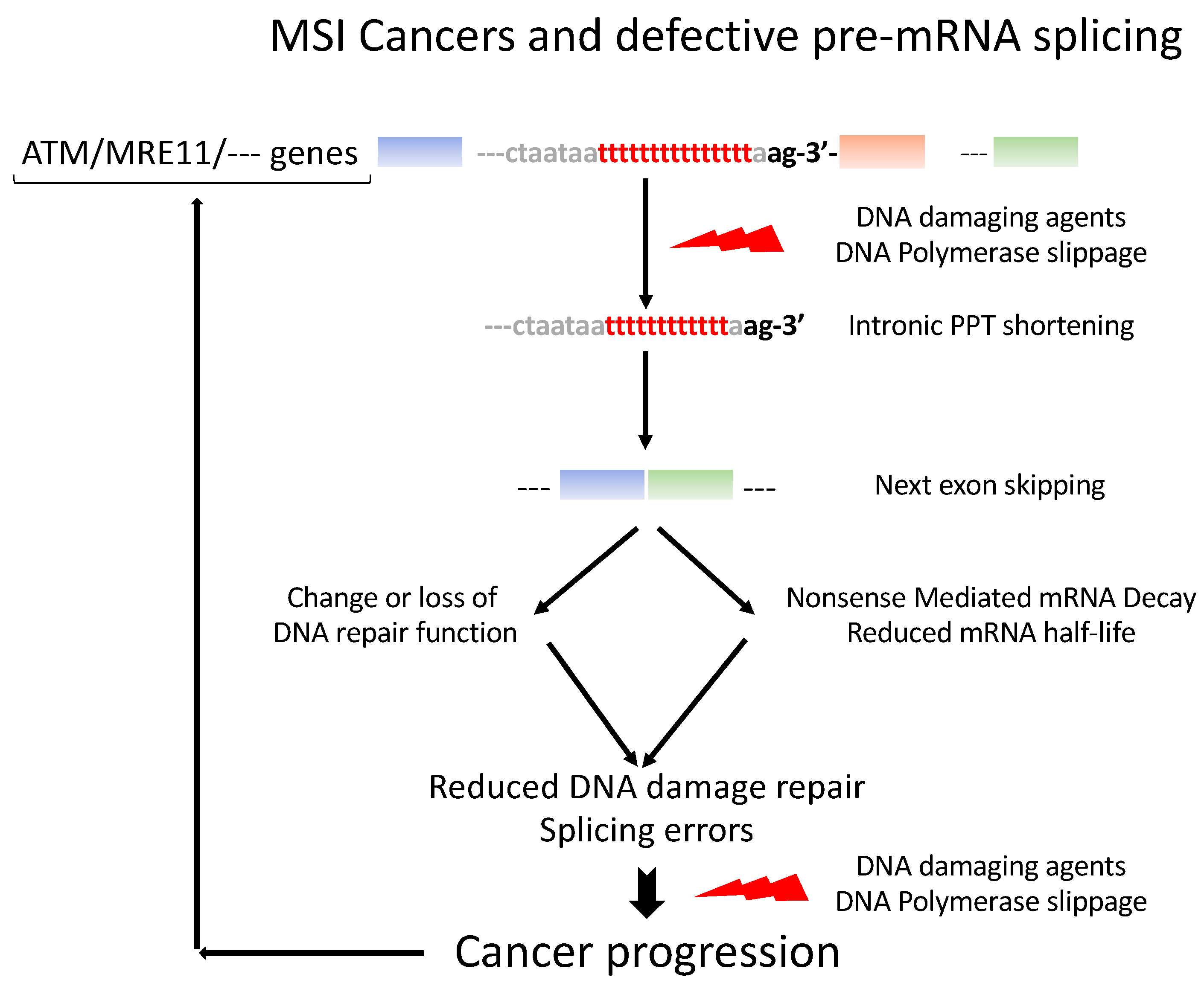
5. Ever-Growing Interplay between Pre-mRNA Splicing and the DNA Damage Response (Figure 3)

6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, G.; Daguenet, É.; Bernard, D.G.; Flodrops, M.; Durand, S.; Chauveau, A.; El Khoury, F.; Le Jossic-Corcos, C.; Corcos, L. L’épissage des ARN pré-messagers: Quand le splicéosome perd pied [Pre-mRNA splicing: When the spliceosome loses ground]. Med. Sci. 2016, 32, 1103–1110. (In French) [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Lansiaux, A.; Logette, E.; Wu, J.; Soret, J.; Tazi, J.; Bailly, C.; Desoche, L.; Solary, E.; Corcos, L. Topoisomerase I and II inhibitors control caspase-2 pre-messenger RNA splicing in human cells. Mol. Cancer Res. 2004, 2, 53–61. [Google Scholar] [CrossRef]
- Boer, R.E.; Torrey, Z.R.; Schneekloth, J.S., Jr. Chemical Modulation of Pre-mRNA Splicing in Mammalian Systems. ACS Chem. Biol. 2020, 15, 808–818. [Google Scholar] [CrossRef]
- Singh, A.; Rajeevan, A.; Gopalan, V.; Agrawal, P.; Day, C.P.; Hannenhalli, S. Broad misappropriation of developmental splicing profile by cancer in multiple organs. Nat. Commun. 2022, 13, 7664. [Google Scholar] [CrossRef]
- Lejeune, F.; Maquat, L. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr. Opin. Cell Biol. 2005, 17, 309–315. [Google Scholar] [CrossRef]
- Dujardin, G.; Kornblihtt, A.R.; Corcos, L. Régulation cinétique de l’épissage alternatif des pré-ARN messagers: Attention au ralentissement [Kinetic regulation of pre-messenger RNA alternative splicing]. Med. Sci. 2014, 30, 940–943. (In French) [Google Scholar] [CrossRef]
- Dujardin, G.; Lafaille, C.; de la Mata, M.; Marasco, L.E.; Muñoz, M.J.; Le Jossic-Corcos, C.; Corcos, L.; Kornblihtt, A. How slow RNA polymerase II elongation favors alternative exon skipping. Mol. Cell 2014, 54, 683–690. [Google Scholar] [CrossRef]
- Pesson, M.; Volant, A.; Uguen, A.; Trillet, K.; De La Grange, P.; Aubry, M.; Daoulas, M.; Robaszkiewicz, M.; Le Gac, G.; Morel, A.; et al. A gene expression and pre-mRNA splicing signature that marks the adenoma-adenocarcinoma progression in colorectal cancer. PLoS ONE 2014, 9, e87761. [Google Scholar] [CrossRef]
- Cilloni, D.; Itri, F.; Bonuomo, V.; Petiti, J. SF3B1 Mutations in Hematological Malignancies. Cancers 2022, 14, 4927. [Google Scholar] [CrossRef]
- Viguera, E.; Canceill, D.; Ehrlich, S. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef]
- Murat, P.; Guilbaud, G.; Sale, J. DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 2020, 21, 209. [Google Scholar] [CrossRef]
- Hamelin, R.; Chalastanis, A.; Colas, C.; El Bchiri, J.; Mercier, D.; Schreurs, A.S.; Simon, V.; Svrcek, M.; Zaanan, A.; Borie, C.; et al. Conséquences cliniques et moléculaires de l’instabilité des microsatellites dans les cancers humains [Clinical and molecular consequences of microsatellite instability in human cancers]. Bull. Cancer 2008, 95, 121–132. (In French) [Google Scholar] [CrossRef]
- Stevens, M.; Oltean, S. Modulation of the Apoptosis Gene Bcl-x Function Through Alternative Splicing. Front. Genet. 2019, 10, 804. [Google Scholar] [CrossRef]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef]
- Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol. Rep. 2016, 35, 1013–1019. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Bonnal, S.; Vigevani, L.; Valcárcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug. Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- De Nicola, A.B.; Tang, Y. Therapeutic approaches to treat human spliceosomal diseases. Curr. Opin. Biotechnol. 2019, 60, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; McCleland, M.L.; Yee, S.; Yaylaoglu, M.; Hussain, S.; Cosino, E.; Quinones, G.; Modrusan, Z.; Seshagiri, S.; Torres, E.; et al. An integrative analysis of colon cancer identifies an essential function for PRPF6 in tumor growth. Genes Dev. 2014, 28, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Taieb, J.; Svrcek, M.; Cohen, R.; Basile, D.; Tougeron, D.; Phelip, J. Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment. Eur. J. Cancer 2022, 175, 136–157. [Google Scholar] [CrossRef]
- Duval, A.; Reperant, M.; Hamelin, R. Comparative analysis of mutation frequency of coding and non coding short mononucleotide repeats in mismatch repair deficient colorectal cancers. Oncogene 2002, 21, 8062–8066. [Google Scholar] [CrossRef]
- Abu-Ghazaleh, N.; Kaushik, V.; Gorelik, A.; Jenkins, M.; Macrae, F. Worldwide prevalence of Lynch syndrome in patients with colorectal cancer: Systematic review and meta-analysis. Genet. Med. 2022, 24, 971–985. [Google Scholar] [CrossRef]
- Ottini, L.; Falchetti, M.; Saieva, C.; De Marco, M.; Masala, G.; Zanna, I.; Paglierani, M.; Giannini, G.; Gulino, A.; Nesi, G.; et al. MRE11 expression is impaired in gastric cancer with microsatellite instability. Carcinogenesis 2004, 25, 2337–2343. [Google Scholar] [CrossRef]
- Ham, M.F.; Takakuwa, T.; Luo, W.J.; Liu, A.; Horii, A.; Aozasa, K. Impairment of double-strand breaks repair and aberrant splicing of ATM and MRE11 in leukemia-lymphoma cell lines with microsatellite instability. Cancer Sci. 2006, 97, 226–234. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Khan, E.S.; Danckwardt, S. Pathophysiological Role and Diagnostic Potential of R-Loops in Cancer and Beyond. Genes 2022, 13, 2181. [Google Scholar] [CrossRef]
- Dorard, C.; de Thonel, A.; Collura, A.; Marisa, L.; Svrcek, M.; Lagrange, A.; Jego, G.; Wanherdrick, K.; Joly, A.L.; Buhard, O.; et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 2011, 17, 1283–1289. [Google Scholar] [CrossRef]
- Collura, A.; Lagrange, A.; Svrcek, M.; Marisa, L.; Buhard, O.; Guilloux, A.; Wanherdrick, K.; Dorard, C.; Taieb, A.; Saget, A.; et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil–based chemotherapy. Gastroenterology 2014, 146, 401–411.e1. [Google Scholar] [CrossRef]
- Hou, W.; Yi, C.; Zhu, H. Predictive biomarkers of colon cancer immunotherapy: Present and future. Front. Immunol. 2022, 13, 1032314. [Google Scholar] [CrossRef]
- Liu, J.; McCleland, M.; Stawiski, E.W.; Gnad, F.; Mayba, O.; Haverty, P.M.; Durinck, S.; Chen, Y.J.; Klijn, C.; Jhunjhunwala, S.; et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat. Commun. 2014, 5, 3830. [Google Scholar] [CrossRef]
- Huiping, C.; Kristjansdottir, S.; Bergthorsson, J.T.; Jonasson, J.G.; Magnusson, J.; Egilsson, V.; Ingvarsson, S. High frequency of LOH, MSI and abnormal expression of FHIT in gastric cancer. Eur. J. Cancer 2002, 38, 728–735. [Google Scholar] [CrossRef]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef]
- Bilbao, C.; Ramírez, R.; Rodríguez, G.; Falcón, O.; León, L.; Díaz-Chico, N.; Perucho, M.; Díaz-Chico, J. Double strand break repair components are frequent targets of microsatellite instability in endometrial cancer. Eur. J. Cancer 2010, 46, 2821–2827. [Google Scholar] [CrossRef]
- Klaric, J.A.; Wüst, S.; Panier, S. New Faces of old Friends: Emerging new Roles of RNA-Binding Proteins in the DNA Double-Strand Break Response. Front. Mol. Biosci. 2021, 8, 668821. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. DNA damage-induced signalling in ataxia-telangiectasia and related syndromes. Radiother. Oncol. 2007, 83, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Redon, C.E.; Guirouilh-Barbat, J.; Smith, S.; Solier, S.; Douarre, C.; Conti, C.; Nakamura, A.J.; Das, B.B.; Nicolas, E.; et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009, 10, 887–893. [Google Scholar] [CrossRef]
- Katzenberger, R.J.; Marengo, M.S.; Wassarman, D. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol. Cell Biol. 2006, 26, 9256–9267. [Google Scholar] [CrossRef]
- Leva, V.; Giuliano, S.; Bardoni, A.; Camerini, S.; Crescenzi, M.; Lisa, A.; Biamonti, G.; Montecucco, A. Phosphorylation of SRSF1 is modulated by replicational stress. Nucleic Acids Res. 2012, 40, 1106–1117. [Google Scholar] [CrossRef]
- Li, X.; Manley, J. New talents for an old acquaintance: The SR protein splicing factor ASF/SF2 functions in the maintenance of genome stability. Cell Cycle 2005, 4, 1706–1708. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef]
- Grote, M.; Wolf, E.; Will, C.L.; Lemm, I.; Agafonov, D.E.; Schomburg, A.; Fischle, W.; Urlaub, H.; Lührmann, R. Molecular architecture of the human Prp19/CDC5L complex. Mol. Cell Biol. 2010, 30, 2105–2119. [Google Scholar] [CrossRef]
- Dellago, H.; Khan, A.; Nussbacher, M.; Gstraunthaler, A.; Lämmermann, I.; Schosserer, M.; Mück, C.; Anrather, D.; Scheffold, A.; Ammerer, G.; et al. ATM-dependent phosphorylation of SNEVhPrp19/hPso4 is involved in extending cellular life span and suppression of apoptosis. Aging 2012, 4, 290–304. [Google Scholar] [CrossRef]
- Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. [Google Scholar] [CrossRef]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corcos, L.; Le Scanf, E.; Quéré, G.; Arzur, D.; Cueff, G.; Jossic-Corcos, C.L.; Le Maréchal, C. Microsatellite Instability and Aberrant Pre-mRNA Splicing: How Intimate Is It? Genes 2023, 14, 311. https://doi.org/10.3390/genes14020311
Corcos L, Le Scanf E, Quéré G, Arzur D, Cueff G, Jossic-Corcos CL, Le Maréchal C. Microsatellite Instability and Aberrant Pre-mRNA Splicing: How Intimate Is It? Genes. 2023; 14(2):311. https://doi.org/10.3390/genes14020311
Chicago/Turabian StyleCorcos, Laurent, Enora Le Scanf, Gaël Quéré, Danielle Arzur, Gwennina Cueff, Catherine Le Jossic-Corcos, and Cédric Le Maréchal. 2023. "Microsatellite Instability and Aberrant Pre-mRNA Splicing: How Intimate Is It?" Genes 14, no. 2: 311. https://doi.org/10.3390/genes14020311
APA StyleCorcos, L., Le Scanf, E., Quéré, G., Arzur, D., Cueff, G., Jossic-Corcos, C. L., & Le Maréchal, C. (2023). Microsatellite Instability and Aberrant Pre-mRNA Splicing: How Intimate Is It? Genes, 14(2), 311. https://doi.org/10.3390/genes14020311