

Characterization and Application of EST-SSR Markers Developed from Transcriptome Sequences in Elymus breviaristatus (Poaceae: Triticeae)


Abstract
1. Introduction
2. Materials and Methods
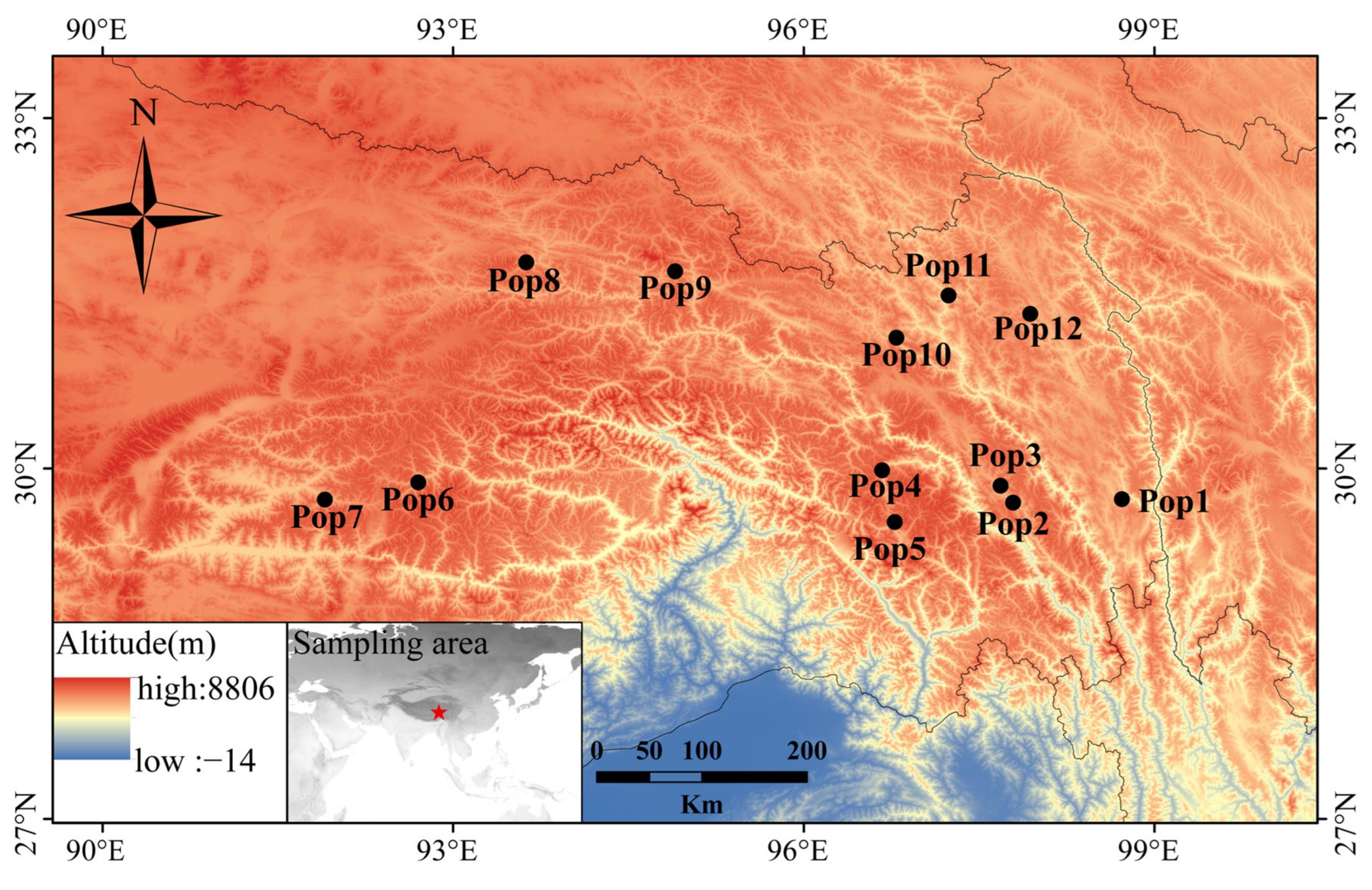
2.1. Plant Materials
2.2. RNA Sequencing, Transcriptome Assembly, and Transcriptome Annotation
2.3. EST-SSR Marker Identification and Primer Design
2.4. DNA Extraction and EST-SSR Amplification
2.5. Statistical Analyses
3. Results
3.1. Illumina Sequencing and De Novo Transcriptome Assembly
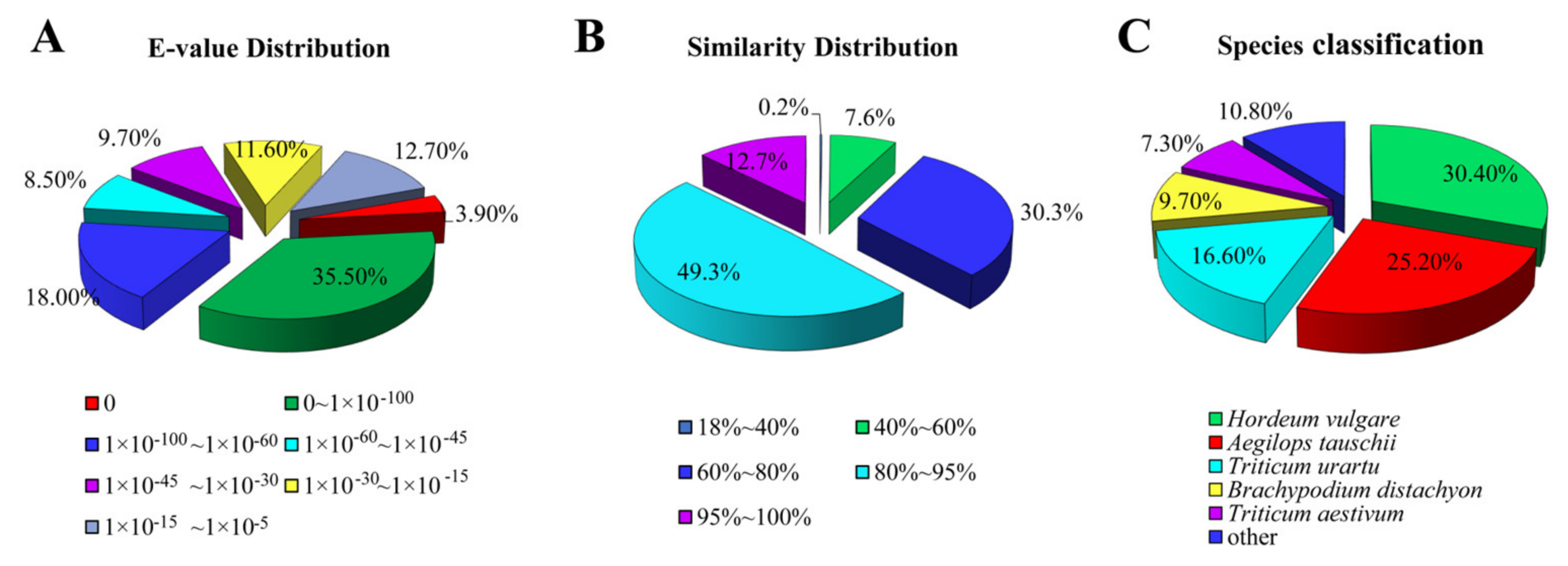
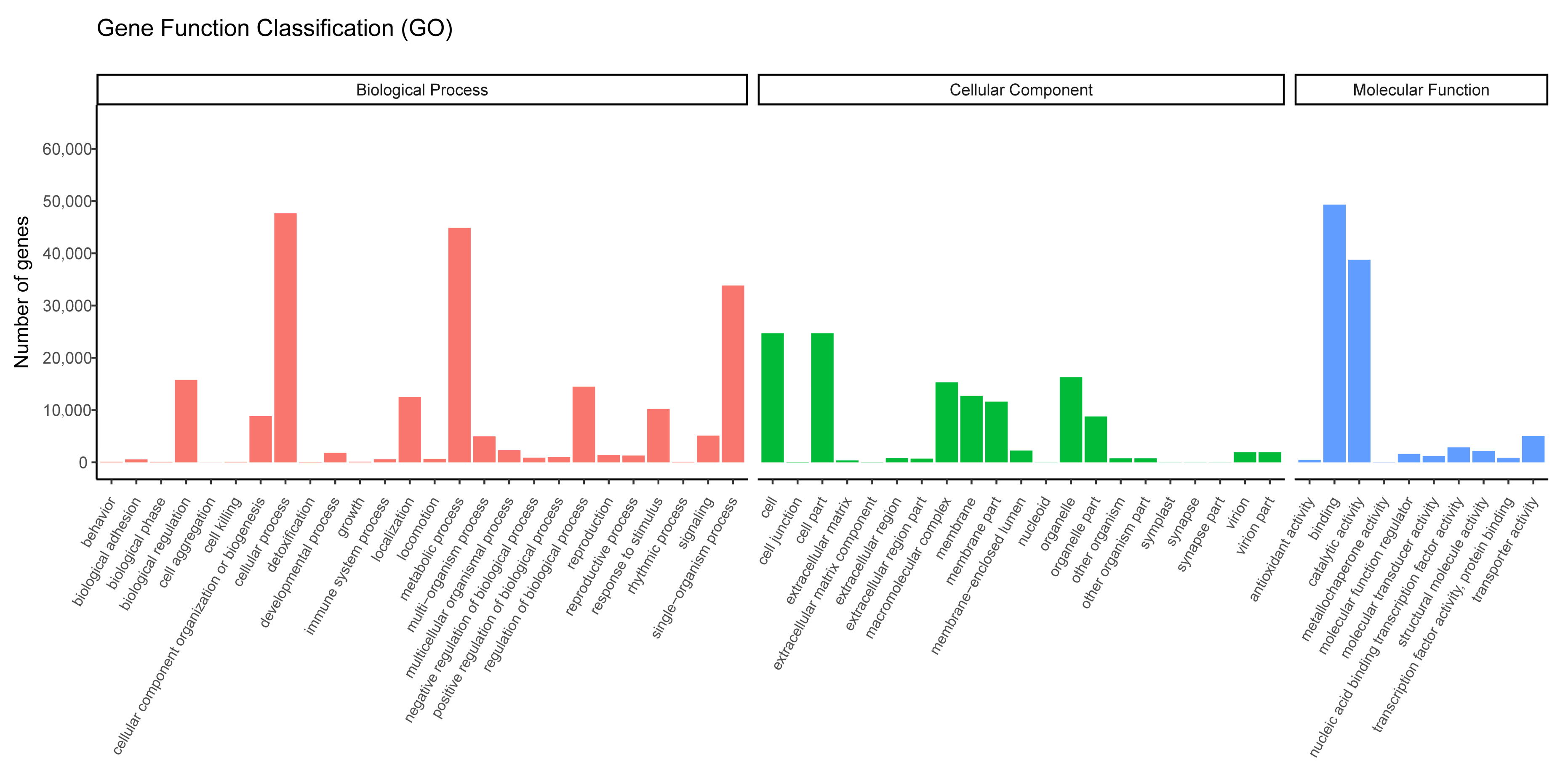
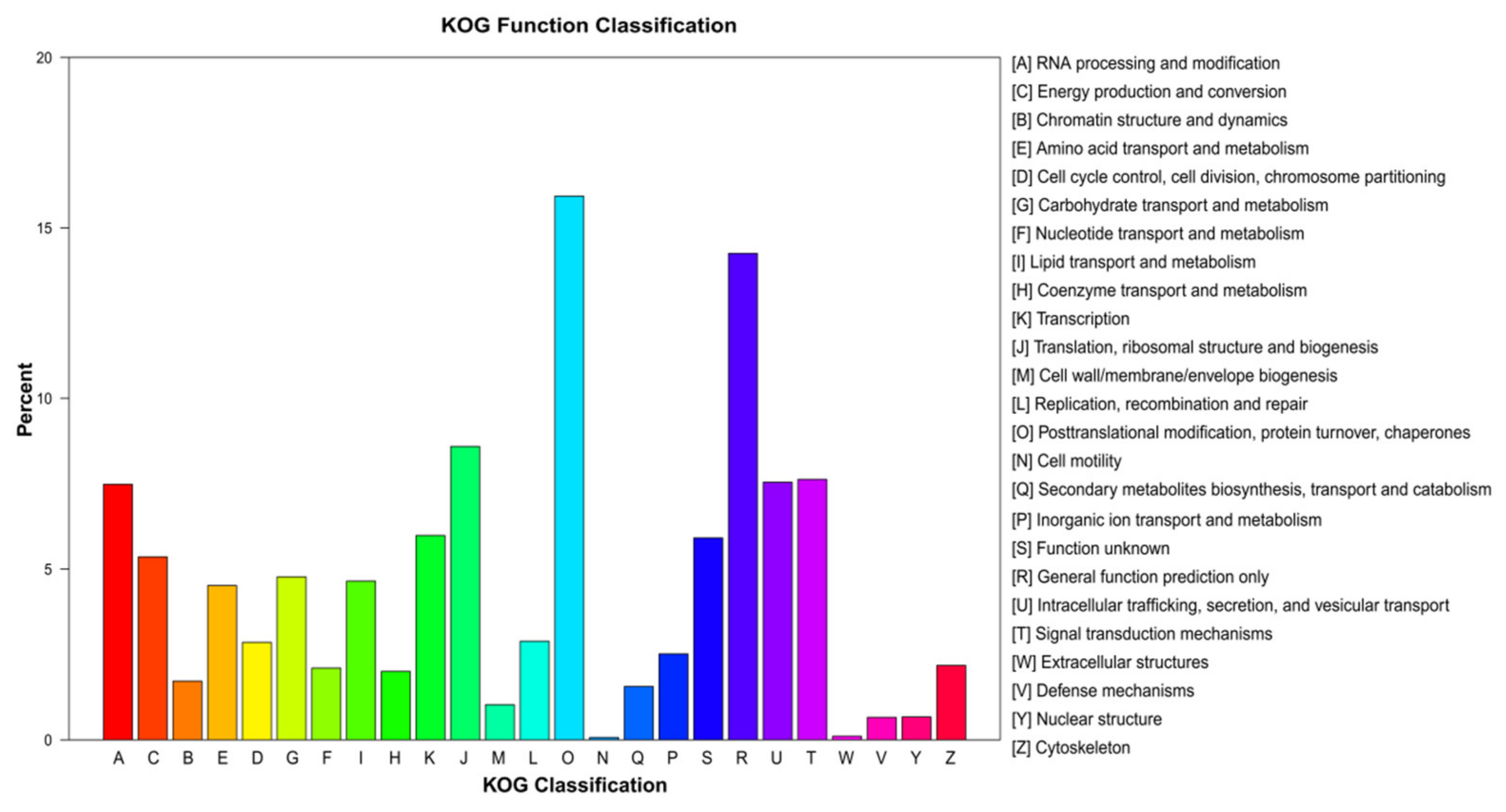
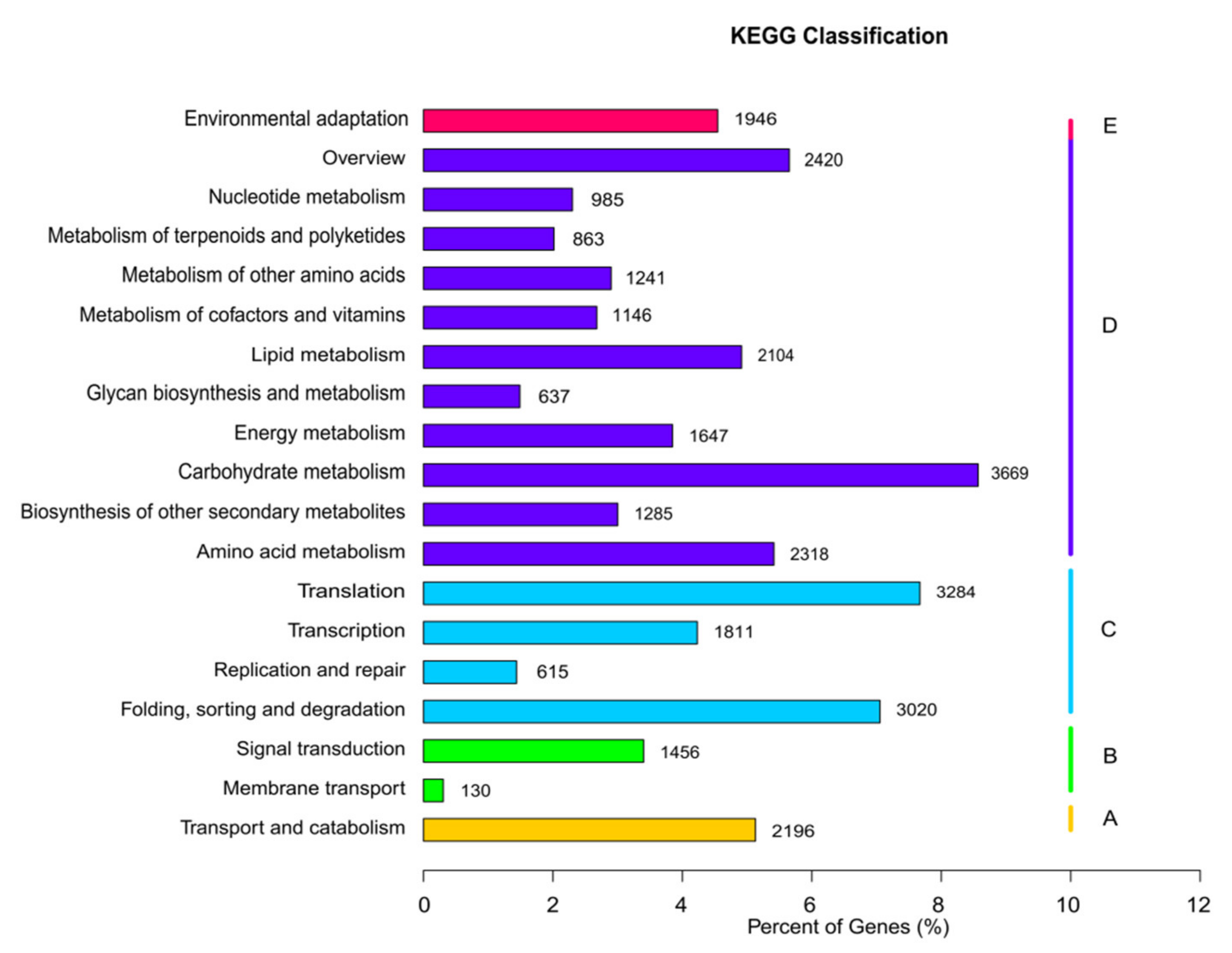
3.2. Unigene Annotation
3.3. Distribution and Frequency of SSR Markers
3.4. Development and Validation of SSR Markers
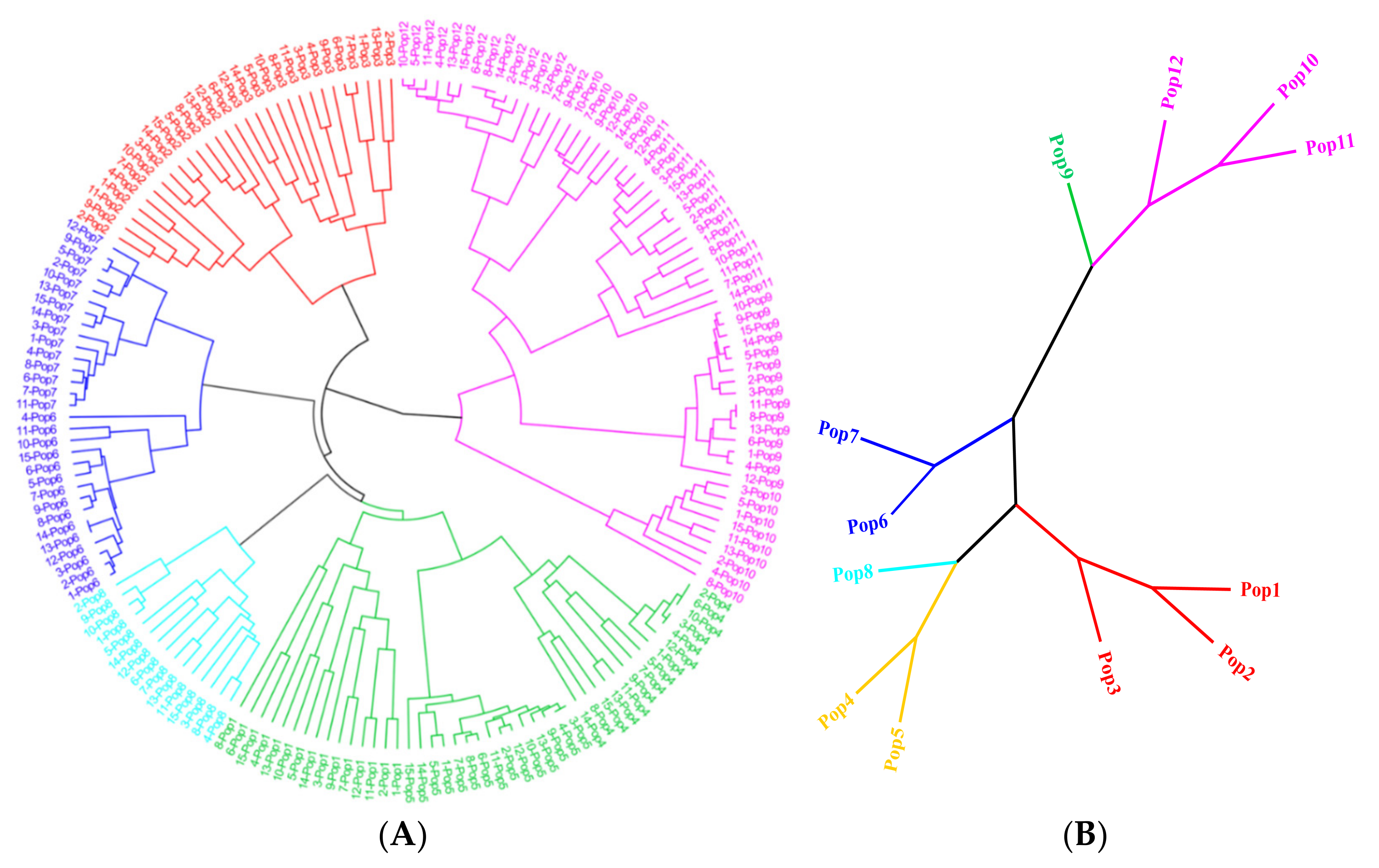
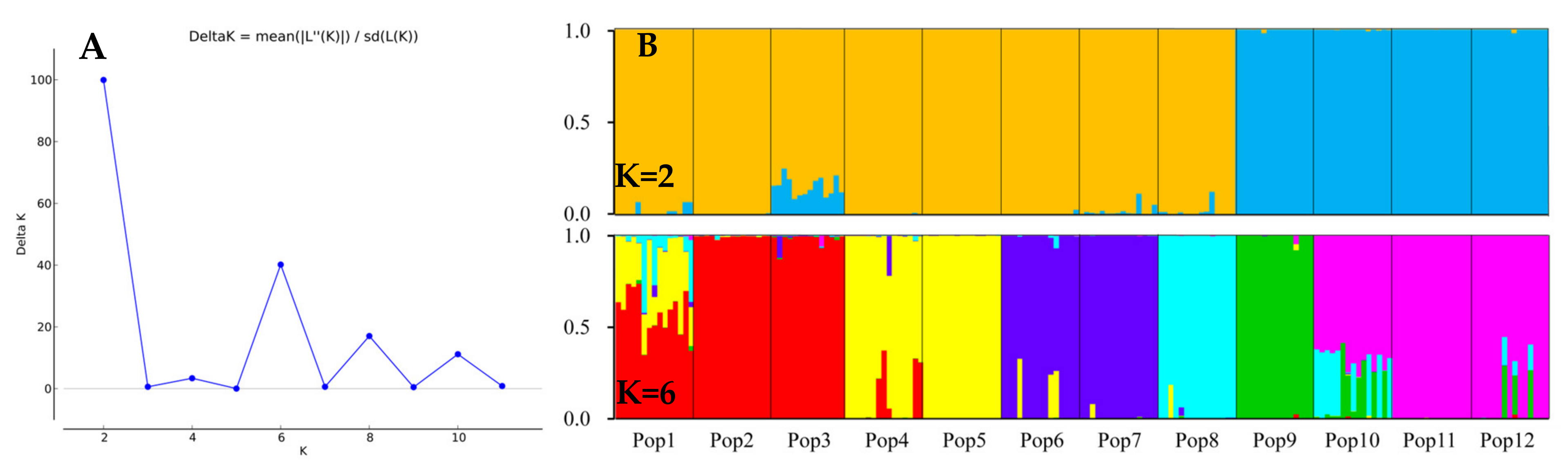
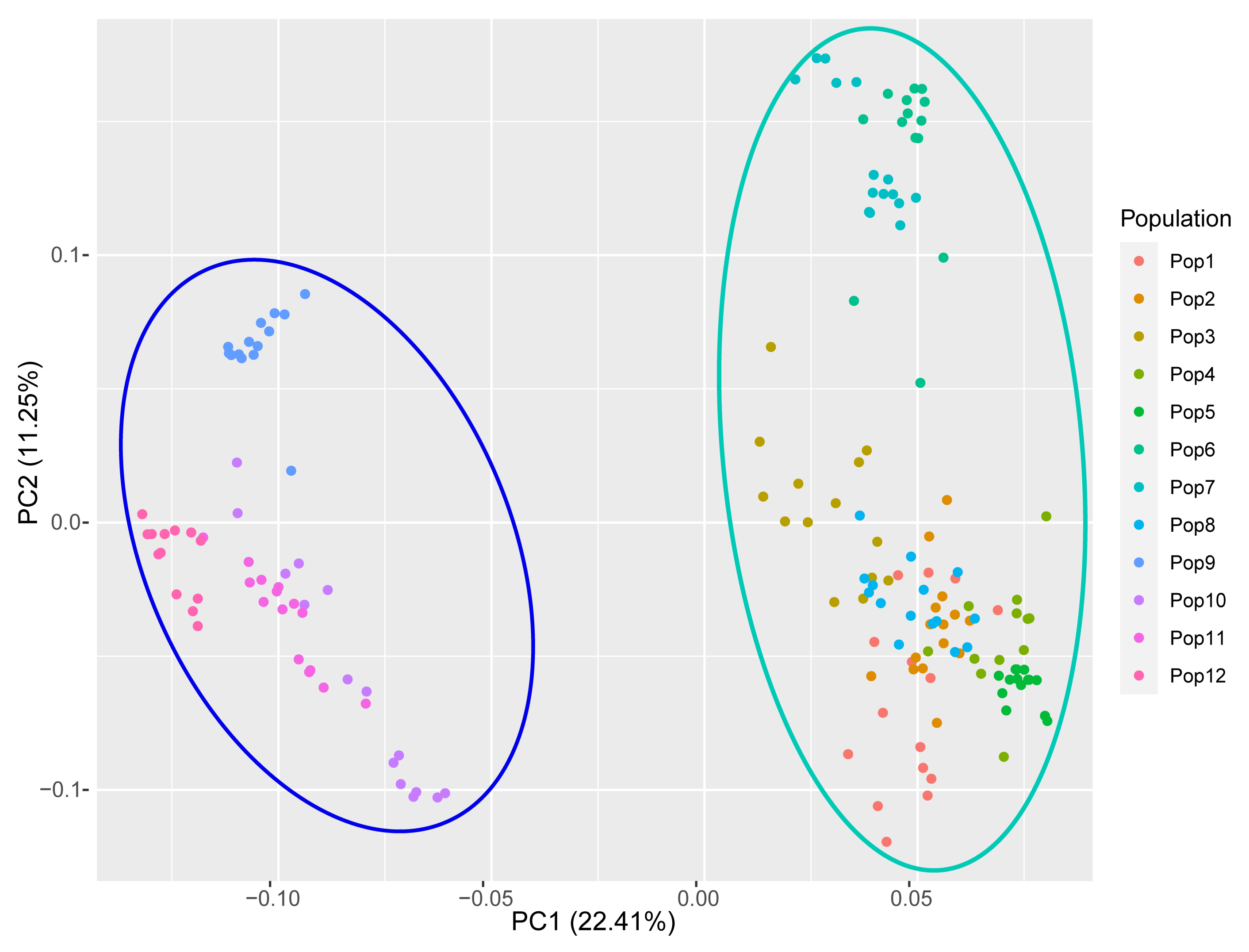
3.5. Population Genetic Structure of E. breviaristatus
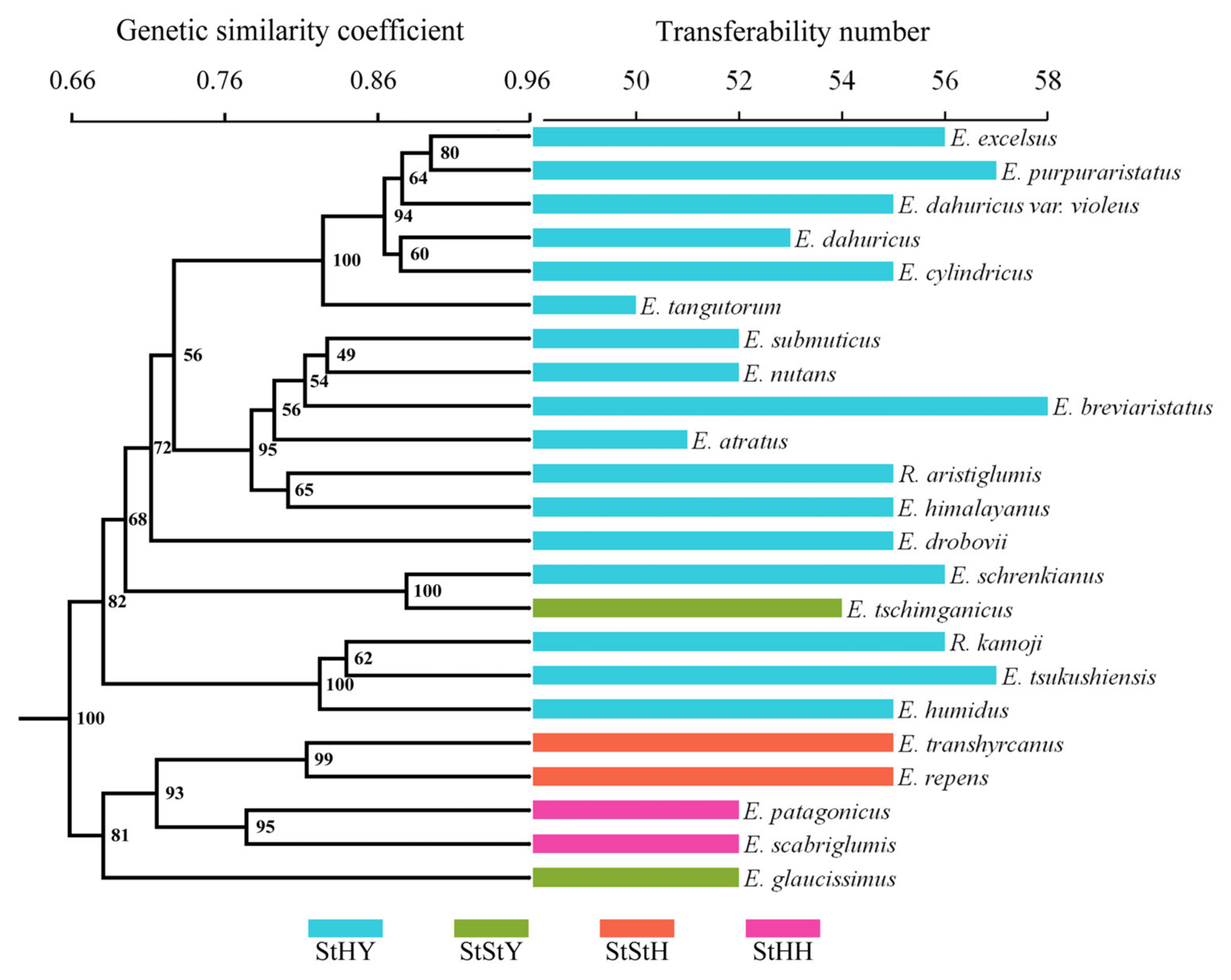
3.6. Transferability of EST-SSRs to Related Species
4. Discussion
4.1. Characterization of SSRs in the Transcriptome
4.2. Genetic Diversity and Structure of E. breviarstatus Populations
4.3. Transferability of EST-SSRs and Phylogenetic Relationships of Elymus Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dewey, D.R. The Genomic System of Classification as a Guide to Intergeneric Hybridization with the Perennial Triticeae; Gustafson, J.P., Ed.; Gene Manipulation in Plant Improvement; Columbia University Press: New York, NY, USA, 1984. [Google Scholar]
- Löve, À. Conspectus of the Triticeae. Feddes Repert. 1984, 95, 425–521. [Google Scholar] [CrossRef]
- Cainong, J.C.; Bockus, W.W.; Feng, Y.; Chen, P.; Qi, L.; Sehgal, S.K.; Danilova, T.V.; Koo, D.; Friebe, B.; Gill, B.S. Chromosome engineering, mapping, and transferring of resistance to Fusarium head blight disease from Elymus tsukushiensis into wheat. Theor. Appl. Genet. 2015, 128, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Keng, Y.L. Flora Illustralis Plantarum Primarum Sinicarum, Gramineae; Science Press: Beijing, China, 1959; pp. 342–399. [Google Scholar]
- Chen, S.L.; Zhu, G.H. Tribus Triticeae, Poaceae; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Flora of China; Science Press: Beijing, China, 2006; pp. 386–444. [Google Scholar]
- Muriira, N.G.; Muchugi, A.; Yu, A.; Xu, J.; Liu, A. Genetic diversity analysis reveals genetic differentiation and strong population structure in Calotropis plants. Sci. Rep. 2018, 8, 7832. [Google Scholar] [CrossRef]
- Khan, F. Molecular markers: An excellent tool for genetic analysis. J. Mol. Biomark. Diagn. 2015, 6, 233. [Google Scholar] [CrossRef]
- Arif, I.A.; Khan, H.A.; Bahkali, A.H.; Al Homaidan, A.A.; Al Farhan, A.H.; Al Sadoon, M.; Shobrak, M. DNA marker technology for wildlife conservation. Saudi J. Biol. Sci. 2011, 18, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, H.R.; Lee, A.J.; Da, E.N.; Chung, K.W. Genetic authentication of cultivars with flower variant types using SSR markers in spring orchid, Cymbidium goeringii. Hortic. Environ. Biotechnol. 2020, 61, 577–590. [Google Scholar] [CrossRef]
- Chikh-Rouhou, H.; Mezghani, N.; Mnasri, S.; Mezghani, N.; Garcés-Claver, A. Assessing the Genetic Diversity and Population Structure of a Tunisian Melon (Cucumis melo L.) Collection Using Phenotypic Traits and SSR Molecular Markers. Agronomy 2021, 11, 1121. [Google Scholar] [CrossRef]
- Sun, M.; Dong, Z.X.; Yang, J.; Wu, W.D.; Zhang, C.L.; Zhang, J.B.; Xiong, Y.; Jia, S.G.; Ma, X. Transcriptomic resources for prairie grass (Bromus catharticus): Expressed transcripts, tissue-specific genes and identification and validation of EST-SSR markers. BMC Plant Biol. 2021, 21, 264. [Google Scholar] [CrossRef]
- Song, Y.P.; Jiang, X.B.; Zhang, M.; Wang, Z.L.; Bo, W.H.; An, X.M.; Zhang, Z.Y. Differences of EST-SSR and genomic-SSR markers in assessing genetic diversity in poplar. For. Stud. China 2012, 14, 1–7. [Google Scholar] [CrossRef]
- Gu, X.Y.; Guo, Z.H.; Ma, X.; Bai, S.Q.; Zhang, X.Q.; Zhang, C.B.; Chen, S.Y.; Peng, Y.; Yan, Y.H.; Huang, L.K. Population genetic variability and structure of Elymus breviaristatus (Poaceae: Triticeae) endemic to Qinghai–Tibetan Plateau inferred from SSR markers. Biochem. Syst. Ecol. 2015, 58, 247–256. [Google Scholar] [CrossRef]
- Yu, Q.Q.; Liu, Q.; Xiong, Y.; Xiong, Y.L.; Dong, Z.X.; Yang, J.; Liu, W.; Ma, X.; Bai, S.Q. Genetic diversity and population divergence of a rare, endemic grass (Elymus breviaristatus) in the southeastern Qinghai-Tibetan Plateau. Sustainability 2019, 11, 5863. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xie, W.G.; Zhao, Y.Q.; Zhang, J.C.; Wang, N.; Ntakirutimana, F.; Yan, J.J.; Wang, Y.R. EST-SSR marker development based on RNA-sequencing of E. sibiricus and its application for phylogenetic relationships analysis of seventeen Elymus species. BMC Plant Biol. 2019, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lei, X.; Bai, S.Q.; Xiong, Y.L.; Liu, W.H.; Wu, W.D.; Yu, Q.Q.; Dong, Z.X.; Yang, J.; Ma, X. Genomic survey sequencing, development and characterization of single- and multi-locus genomic SSR markers of Elymus sibiricus L. BMC Plant Biol. 2021, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Abdullah, T.L.; Rafii, M.Y.; Harikrishna, J.A.; Werbrouck, S.P.O.; Teo, C.H.; Sahebi, M.; Azizi, P. De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci. Rep. 2019, 9, 3047. [Google Scholar] [CrossRef]
- Karan, M.; Evans, D.S.; Reilly, D.; Schulte, K.; Wright, C.; Innes, D.; Holton, T.A.; Nikles, D.G.; Dickinson, G.R. Rapid microsatellite marker development for African mahogany (Khaya senegalensis, Meliaceae) using next-generation sequencing and assessment of its intra-specific genetic diversity. Mol. Ecol. Resour. 2012, 12, 344–353. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Yeh, F.C. Population genetic analysis of codominant and dominant markers and quantitative traits. Belg. J. Bot. 1997, 130, 129–157. [Google Scholar]
- Roldán-Ruiz, I.; Dendauw, J.; Bockstaele, E.V.; Depicker, A.; Loose, M.D. AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp.). Mol. Breed. 2000, 6, 125–134. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Microcomputer programs for numerical taxonomy and multivariate analysis. Am. Stat. 1987, 41, 330. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Boil. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Bushman, B.S.; Larson, S.R.; Mott, I.W.; Cliften, P.F.; Wang, R.R.C.; Chatterton, N.J.; Hernandez, A.G.; Ali, S.; Kim, R.W.; Thimmapuram, J.; et al. Development and annotation of perennial Triticeae ESTs and SSR markers. Genome 2008, 51, 779–788. [Google Scholar] [CrossRef]
- Mott, I.W.; Larson, S.R.; Bushman, B.S. Simple sequence repeat (SSR) markers for Elymus, Pseudoroegneria and Pascopyrum species (Triticeae: Gramineae). Plant Genet. Resour. 2011, 9, 489–494. [Google Scholar] [CrossRef]
- Sun, G.L.; Salomon, B.; Von, B.R. Characterization of microsatellite loci from Elymus alaskanus and length polymorphism in several Elymus species (Triticeae: Poaceae). Genome 1998, 41, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Zhang, P.F.; Song, M.L.; Hou, J.L.; Qing, M.; Wang, W.Q.; Liu, C.S.; Chen, S.L. Transcriptome analysis and development of SSR molecular markers in Glycyrrhiza uralensis fisch. PLoS ONE 2015, 10, e0143017. [Google Scholar] [CrossRef]
- Wang, S.F.; Wang, X.F.; He, Q.W.; Liu, X.X.; Xu, W.L.; Li, L.B.; Gao, J.W.; Wang, F.D. Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST-SSR markers in radish. Plant Cell Rep. 2012, 31, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Hiz, M.C.; Canher, B.; Niron, H.; Turet, M. Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS ONE. 2014, 9, e92598. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Zhou, Q.; Ma, L.C.; Xie, W.G.; Wang, Y.R.; Hu, X.W.; Liu, Z.P. Novel polymorphic expressed–sequence tag–simple–sequence repeat markers in Campeiostachys nutans for genetic diversity analyses. Crop Sci. 2015, 55, 2712–2718. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, H.W.; Fu, X.N.; Li, X.H.; Gao, H.W. Development of simple sequence repeat markers and diversity analysis in alfalfa (Medicago sativa L.). Mol. Biol. Rep. 2013, 40, 3291–3298. [Google Scholar] [CrossRef]
- Xin, D.W.; Sun, J.Y.; Wang, J.L.; Jiang, H.W.; Hu, G.H.; Liu, C. Identification and characterization of SSRs from soybean (Glycine max) ESTs. Mol. Biol. Rep. 2012, 39, 9047–9057. [Google Scholar] [CrossRef]
- Eujayl, I.; Sledge, M.K.; Wang, L.; May, G.D.; Chekhovskiy, K.; Zwonitzer, J.C.; Mian, M.A.R. Medicago truncatula EST-SSRs reveal cross-species geneticmarkers for Medicago spp. Theor. Appl. Genet. 2004, 108, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Y.; Liu, Z.; Zhu, X.W.; Zhai, L.L.; Xu, L.; Yu, R.G.; Gong, Y.Q.; Liu, L.W. De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genom. 2013, 14, 836. [Google Scholar] [CrossRef] [PubMed]
- Hina, F.; Yisilam, G.; Wang, S.Y.; Li, P.; Fu, C.X. De novo transcriptome assembly, gene annotation and SSR marker development in the moon seed genus Menispermum (Menispermaceae). Front. Genet. 2020, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Thiel, T.; Stein, N.; Langridge, P.; Graner, A. In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell. Mol. Biol. Lett. 2002, 7, 537–546. [Google Scholar]
- You, Y.N.; Liu, D.C.; Liu, H.B.; Zheng, X.F.; Diao, Y.; Huang, X.F.; Hu, Z.L. Development and characterisation of EST-SSR markers by transcriptome sequencing in taro (Colocasia esculenta (L.) Schoot). Mol. Breed. 2015, 35, 134. [Google Scholar] [CrossRef]
- Wu, J.; Cai, C.F.; Cheng, F.Y.; Cui, H.L.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Kuleung, C.; Baenziger, P.S.; Dweikat, I. Transferability of SSR markers among wheat, rye, and triticale. Theor. Appl. Genet. 2004, 108, 1147–1150. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef]
- Sanders, T.B.; Hamrick, J.L.; Holden, L.R. Allozyme variation in Elymus canadensis from the tallgrass prairie region: Geographic variation. Am. Midl. Nat. 1979, 101, 1–12. [Google Scholar] [CrossRef]
- Díaz, O.; Sun, G.L.; Salomon, B.; Von Bothmer, R. Levels and distribution of allozyme and RAPD variation in populations of Elymus fibrosus (Schrenk) Tzvel. (Poaceae). Genet. Resour. Crop Evol. 2000, 7, 11–24. [Google Scholar] [CrossRef]
- Sun, G.L.; Diaz, O.; Salomon, B.; Von Bothmer, R. Genetic diversity and structure in a natural Elymus caninus population from Denmark based on microsatellite and isozyme analyses. Plant Syst. Evol. 2001, 227, 235–244. [Google Scholar] [CrossRef]
- Yan, X.B.; Guo, Y.X.; Zhou, H.; Lu, B.R.; Wang, K. Genetic patterns of ten Elymus species from the Tibetan and Inner Mongolian plateaus of China. Grass Forage Sci. 2006, 61, 398–404. [Google Scholar] [CrossRef]
- Brzosko, E.; Ratkiewicz, M.; Wróblewska, A. Allozyme differentiation and genetic structure of the Lady’s slipper (Cypripedium calceolus) island populations in north-east Poland. Bot. J. Linn. Soc. 2015, 138, 433–440. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, D.; Ma, L.C.; Xie, W.G.; Wang, Y.; Wang, Y.R.; Liu, Z.P. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6, 20549. [Google Scholar] [CrossRef]
- Chen, S.Y.; Ma, X.; Zhang, X.Q.; Chen, Z.H. Karyotypes of 10 tetraploid species in Elymus (Poaceae: Triticeae). J. Syst. Evol. 2008, 46, 886–890. [Google Scholar]
- Baum, B.R.; Yen, C.; Yang, J.L. Taxonomic separation of Kengyilia (Poaceae: Triticeae) in relation to nearest related Roegneria, Elymus, and Agropyron, based on some morphological characters. Plant Syst. Evol. 1995, 194, 123–132. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range (bp) | Transcripts | Unigene |
|---|---|---|
| 200–500 | 106,453 | 36,753 |
| 500–1000 | 67,045 | 59,577 |
| 1000–2000 | 57,272 | 55,935 |
| >2000 | 19,412 | 19,257 |
| Total Number | 250,182 | 171,522 |
| Total Length | 215,188,383 | 187,855,868 |
| N50 Length | 1268 | 1409 |
| N90 Length | 377 | 570 |
| Mean Length | 860 | 1095 |
| Number of Repeat Units | Mono- | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Percentage (%) |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 8687 | 505 | 119 | 54 | 9365 | 30.54 |
| 6 | 0 | 2263 | 3544 | 166 | 5 | 17 | 5995 | 19.55 |
| 7 | 0 | 961 | 1360 | 22 | 7 | 1 | 2351 | 7.67 |
| 8 | 0 | 668 | 560 | 12 | 1 | 0 | 1241 | 4.05 |
| 9 | 0 | 537 | 88 | 3 | 0 | 0 | 628 | 2.05 |
| 10 | 3616 | 285 | 96 | 0 | 0 | 0 | 3997 | 13.03 |
| 11 | 1460 | 300 | 27 | 1 | 0 | 0 | 1788 | 5.83 |
| 12 | 840 | 307 | 17 | 2 | 0 | 0 | 1166 | 3.80 |
| 13 | 481 | 107 | 22 | 0 | 0 | 0 | 610 | 1.99 |
| 14 | 402 | 126 | 5 | 0 | 0 | 0 | 533 | 1.74 |
| 15 | 332 | 138 | 1 | 0 | 0 | 0 | 471 | 1.54 |
| >15 | 1965 | 549 | 9 | 0 | 0 | 0 | 2797 | 9.12 |
| Total | 9096 | 6241 | 14,416 | 711 | 132 | 72 | 30,668 | 100 |
| Percentage (%) | 29.66 | 20.35 | 47.01 | 2.32 | 0.43 | 0.23 | 100 |
| Primers | Primer Sequence (5′-3′) | Repeat Motif | Fragment Size (bp) | PIC | Mean H | Mean I |
|---|---|---|---|---|---|---|
| MDEB002 | F: AGTCCTTAGCCCCTAGGACG | (CT) 9 | 204–246 | 0.499 | 0.271 | 0.422 |
| R: AAAGAGAGGAGGGCTGGGAT | ||||||
| MDEB024 | F: ATGTATGGGGCGGGCATATG | (CAT) 5 | 203–224 | 0.500 | 0.125 | 0.208 |
| R: CAAACCGACGAGAGGTGTCA | ||||||
| MDEB025 | F: GATCCGACGACCTCAGCTTC | (CCA) 5 | 197–228 | 0.487 | 0.316 | 0.466 |
| R: CCATCTGCAGCTGGTCAAGA | ||||||
| MDEB027 | F: CATCGACATCACCTGGGTGT | (CCG) 5 | 227–241 | 0.498 | 0.275 | 0.414 |
| R: TTCTTGTTCCCCACGGCTTC | ||||||
| MDEB046 | F: TACATCAAAGCGCAGGCAGA | (CTA) 6 | 176–267 | 0.497 | 0.316 | 0.469 |
| R: AAGGAGTTGACATGGCTCGG | ||||||
| MDEB050 | F: CGCCTACACAAGATGGTGGT | (CTG) 5 | 172–257 | 0.499 | 0.425 | 0.612 |
| R: CACTCTGTCGAAATGCAGCG | ||||||
| MDEB051 | F: GCCTGCTGAAGATGCTCAGA | (CTG) 5 | 148–237 | 0.451 | 0.387 | 0.571 |
| R: CCAGCAGTACAACCAGAGCA | ||||||
| MDEB054 | F: ACCGGCATTTCCTACACTGG | (GAC) 6 | 226–235 | 0.419 | 0.304 | 0.475 |
| R: TCCTCACAGCTCTCACCACA | ||||||
| MDEB063 | F: CACTTCTCCCCTCGCGATG | (GCG) 5 | 240–277 | 0.500 | 0.410 | 0.588 |
| R: AACATGAGCAGGCTCTCGAC | ||||||
| MDEB065 | F: CAAGAGCGACCTGGTGAAGT | (GCG) 5 | 156–230 | 0.469 | 0.316 | 0.481 |
| R: CTCGTTCCCATCATCCGCAA | ||||||
| MDEB067 | F: AATGGTTCGTTGTCGTCGGA | (GCG) 6 | 211–219 | 0.496 | 0.301 | 0.453 |
| R: GTGGTGGACAAGTAGCTGCT | ||||||
| MDEB068 | F: GTAACTGTTCTTCGGCGGGA | (GCT) 5 | 145–231 | 0.399 | 0.363 | 0.530 |
| R: CCACCGAAGAACGACTACCC | ||||||
| MDEB076 | F: ATCCACGTTTCCCTCTGCTG | (GGC) 5 | 229–248 | 0.487 | 0.400 | 0.586 |
| R: CCAGTCCCCACTACCAGCTA | ||||||
| MDEB083 | F: TGAGGCTGGGACTGAAGAGT | (GTC) 5 | 191–212 | 0.443 | 0.239 | 0.335 |
| R: CTTCTCAGCTGCTGACCTCC | ||||||
| MDEB091 | F: ACCCCCAGCTACTACCATGT | (TCA) 6 | 226–248 | 0.267 | 0.155 | 0.250 |
| R: GAGATGGAGCCTACCGAAGC | ||||||
| MDEB094 | F: CGCCTCTTCCACGTCTTTGA | (TCG) 5 | 138–267 | 0.493 | 0.294 | 0.443 |
| R: GATGGTGCTCCTCGAGATCG | ||||||
| MDEB095 | F: CAGCTGTTCTGGAGGGTGAG | (TCT) 5 | 216–280 | 0.355 | 0.162 | 0.258 |
| R: TCAGAAGGTGACGACGCTTC | ||||||
| MDEB100 | F: TGGTCGCGAGATCTTATGGG | (TGT) 5 | 184–193 | 0.475 | 0.248 | 0.411 |
| R: TAAACAACAGCACCTGGCCT |
| Pop ID | N | Na | Ne | I | He | uHe |
|---|---|---|---|---|---|---|
| Pop1 | 15 | 12.000 | 11.400 | 0.237 | 0.153 | 0.159 |
| Pop2 | 15 | 8.167 | 10.662 | 0.143 | 0.097 | 0.100 |
| Pop3 | 14 | 9.000 | 10.965 | 0.175 | 0.118 | 0.123 |
| Pop4 | 15 | 8.500 | 10.754 | 0.154 | 0.104 | 0.107 |
| Pop5 | 15 | 5.611 | 9.513 | 0.043 | 0.028 | 0.028 |
| Pop6 | 15 | 7.667 | 10.246 | 0.111 | 0.074 | 0.077 |
| Pop7 | 15 | 6.389 | 9.950 | 0.081 | 0.054 | 0.056 |
| Pop8 | 15 | 8.889 | 10.837 | 0.164 | 0.110 | 0.114 |
| Pop9 | 15 | 6.333 | 9.733 | 0.059 | 0.039 | 0.041 |
| Pop10 | 15 | 8.389 | 10.902 | 0.175 | 0.117 | 0.121 |
| Pop11 | 15 | 7.167 | 10.202 | 0.107 | 0.071 | 0.073 |
| Pop12 | 15 | 6.444 | 9.881 | 0.078 | 0.050 | 0.052 |
| Mean | 7.880 | 10.42 | 0.127 | 0.085 | 0.088 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, C.; Chen, S.; Jiang, K.; Guan, H.; Liu, W. Characterization and Application of EST-SSR Markers Developed from Transcriptome Sequences in Elymus breviaristatus (Poaceae: Triticeae). Genes 2023, 14, 302. https://doi.org/10.3390/genes14020302
Li J, Zhang C, Chen S, Jiang K, Guan H, Liu W. Characterization and Application of EST-SSR Markers Developed from Transcriptome Sequences in Elymus breviaristatus (Poaceae: Triticeae). Genes. 2023; 14(2):302. https://doi.org/10.3390/genes14020302
Chicago/Turabian StyleLi, Jin, Changbing Zhang, Shiyong Chen, Keke Jiang, Hao Guan, and Wenhui Liu. 2023. "Characterization and Application of EST-SSR Markers Developed from Transcriptome Sequences in Elymus breviaristatus (Poaceae: Triticeae)" Genes 14, no. 2: 302. https://doi.org/10.3390/genes14020302
APA StyleLi, J., Zhang, C., Chen, S., Jiang, K., Guan, H., & Liu, W. (2023). Characterization and Application of EST-SSR Markers Developed from Transcriptome Sequences in Elymus breviaristatus (Poaceae: Triticeae). Genes, 14(2), 302. https://doi.org/10.3390/genes14020302