

Parallel Evolution of Sex-Linked Genes across XX/XY and ZZ/ZW Sex Chromosome Systems in the Frog Glandirana rugosa
 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Frogs
2.2. RNA-Sequencing, De Novo Assembly, and Gene Annotation
2.3. Evolutionary Analyses
2.4. Phylogenetic Analyses of Sex Chromosome-Linked Sox3 and tl Genes
2.5. Expression Analyses
3. Results
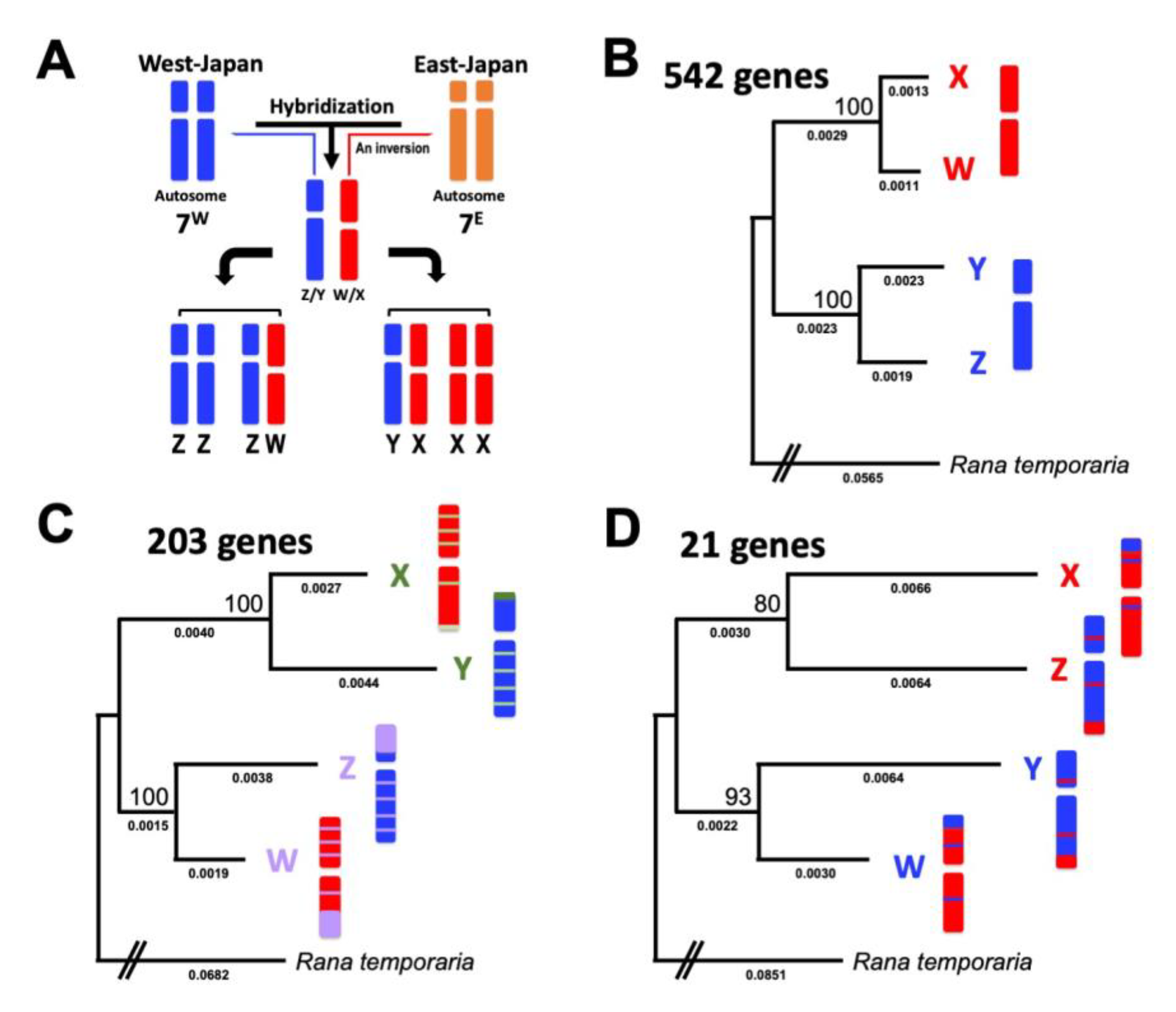
3.1. Identification of X, Y, Z, and W Sex Chromosomes-Linked Genes

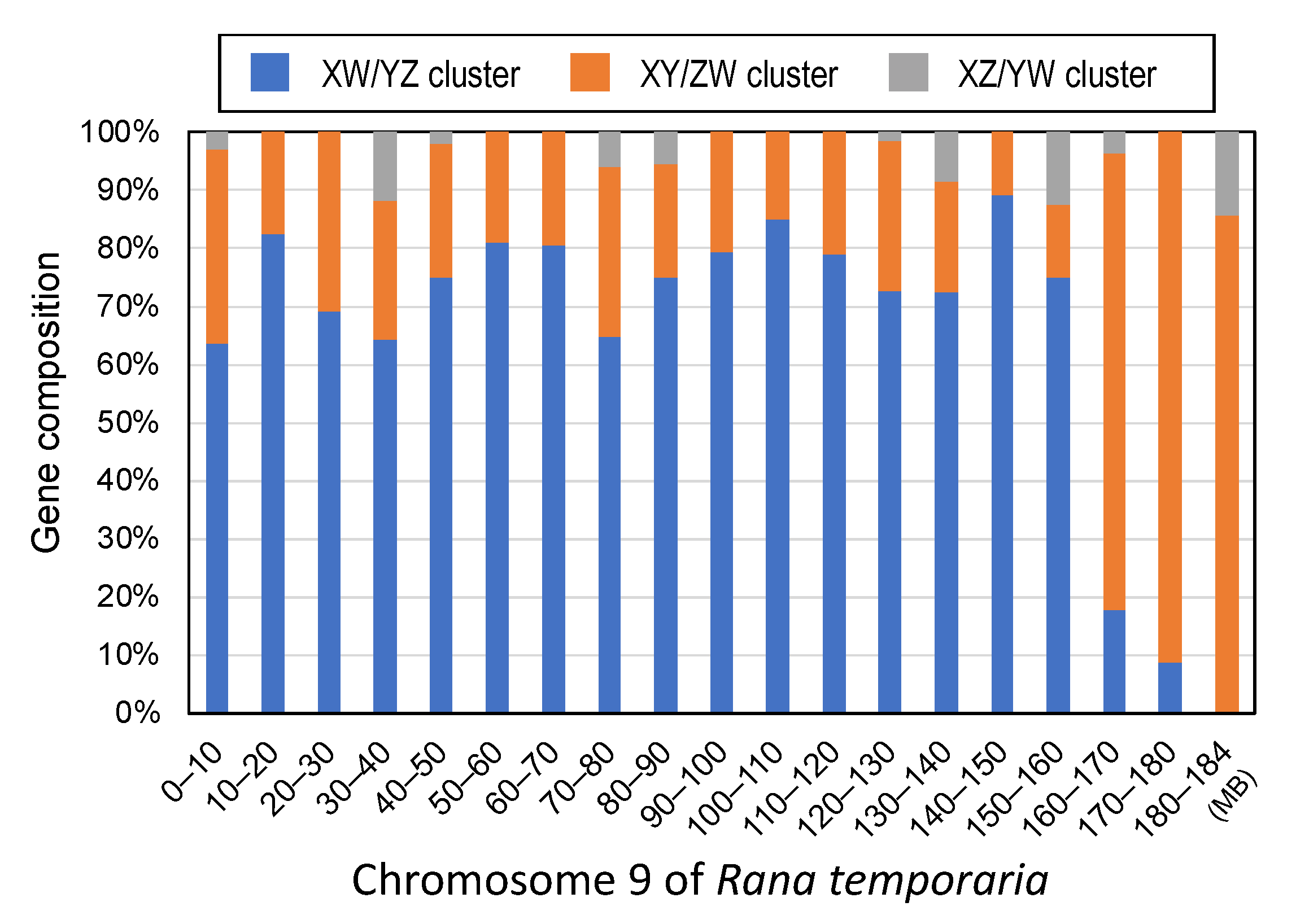
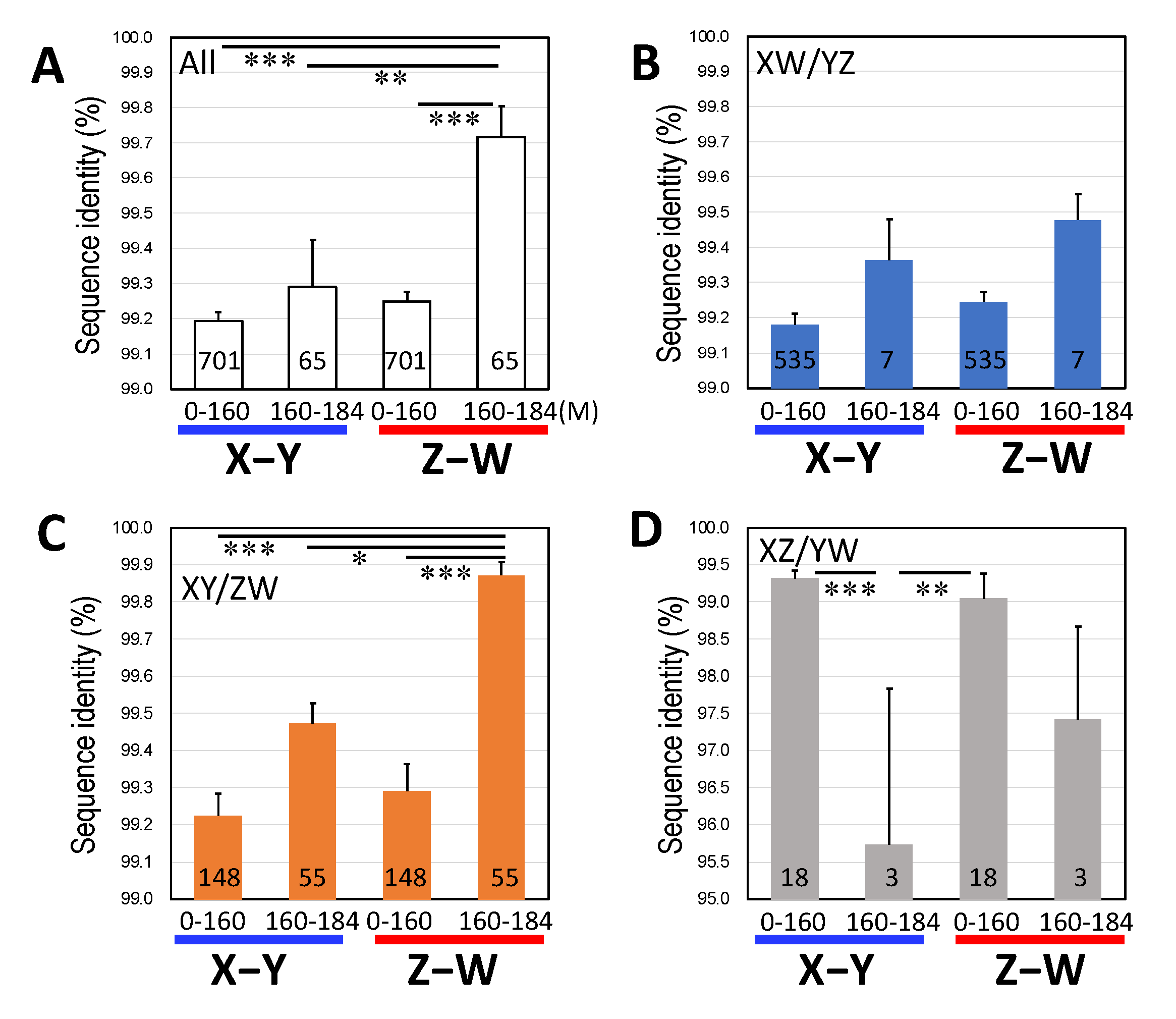
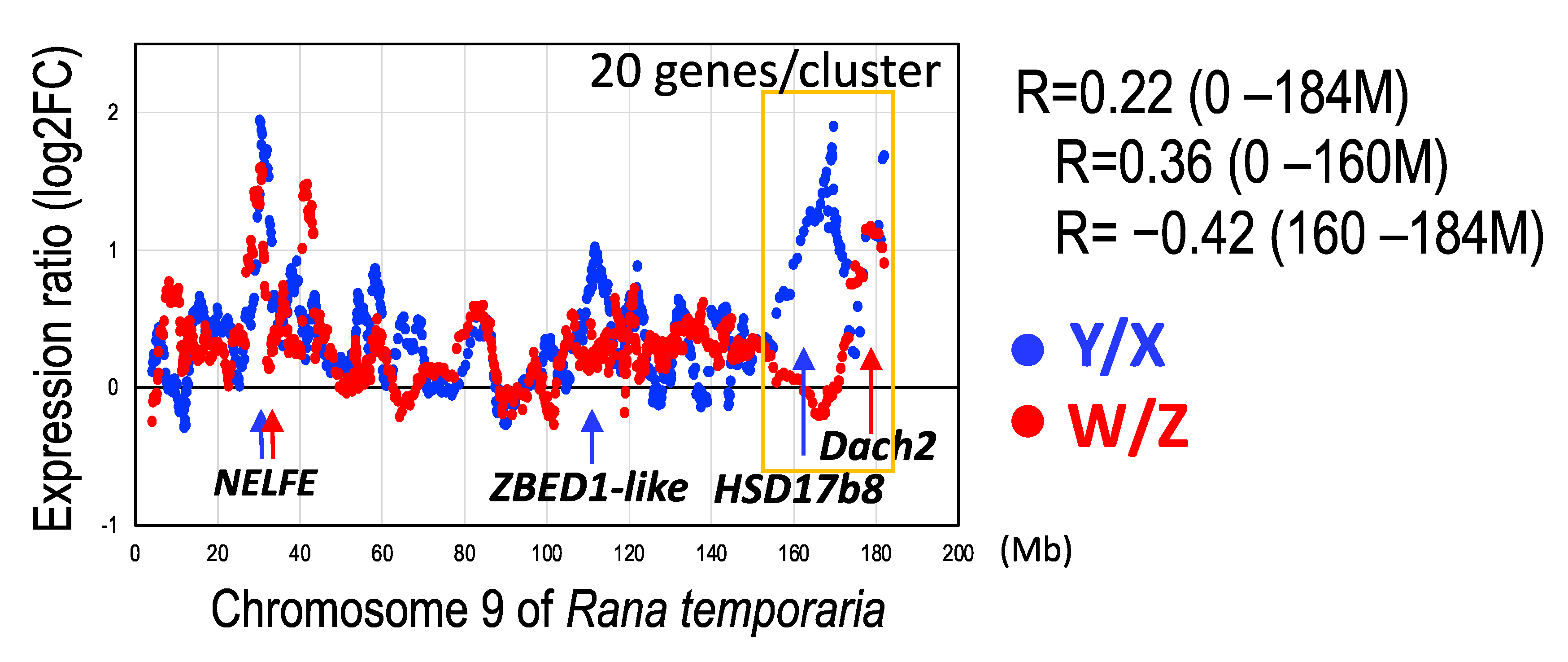
3.2. Three Clusters of Sex-Linked Genes
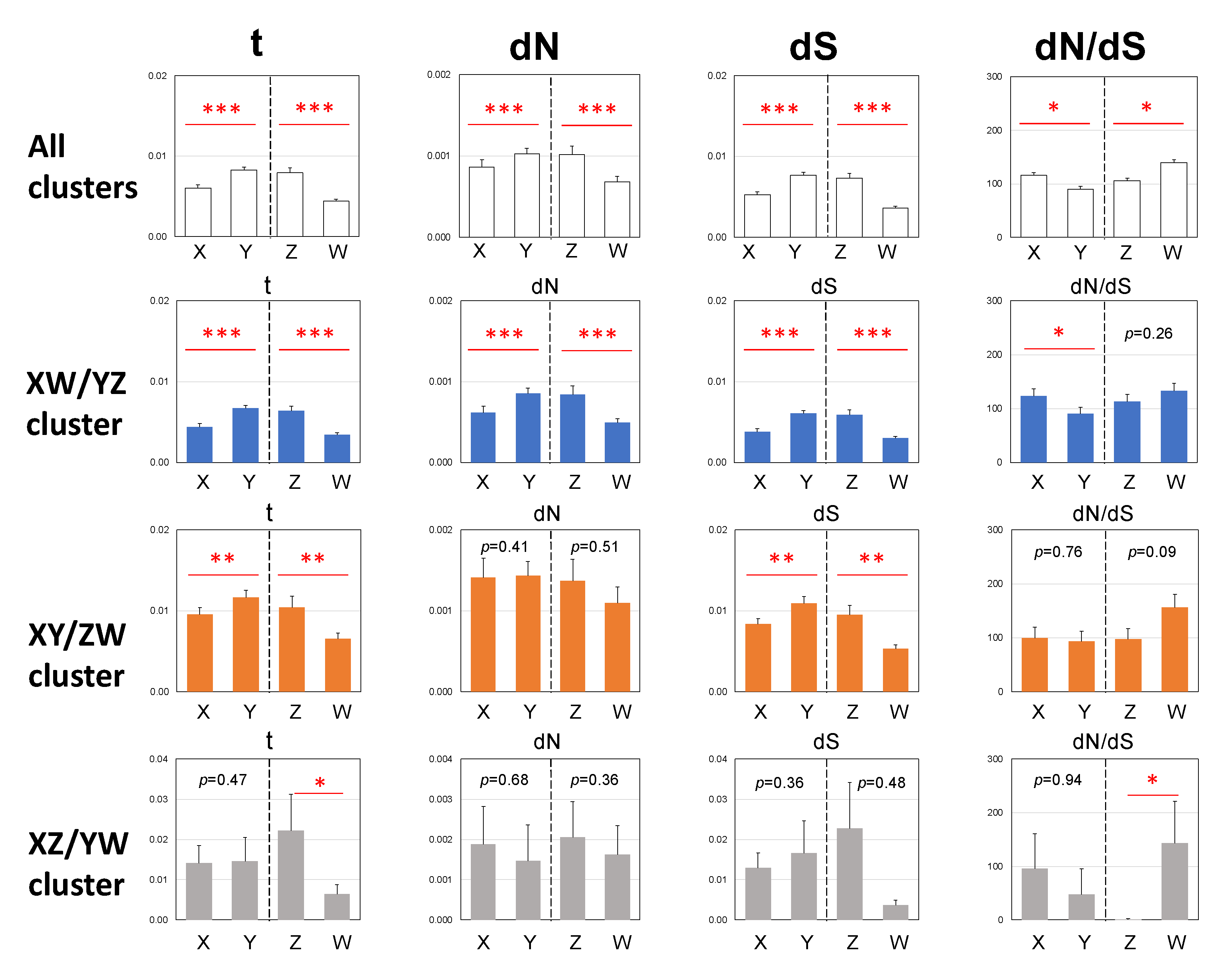
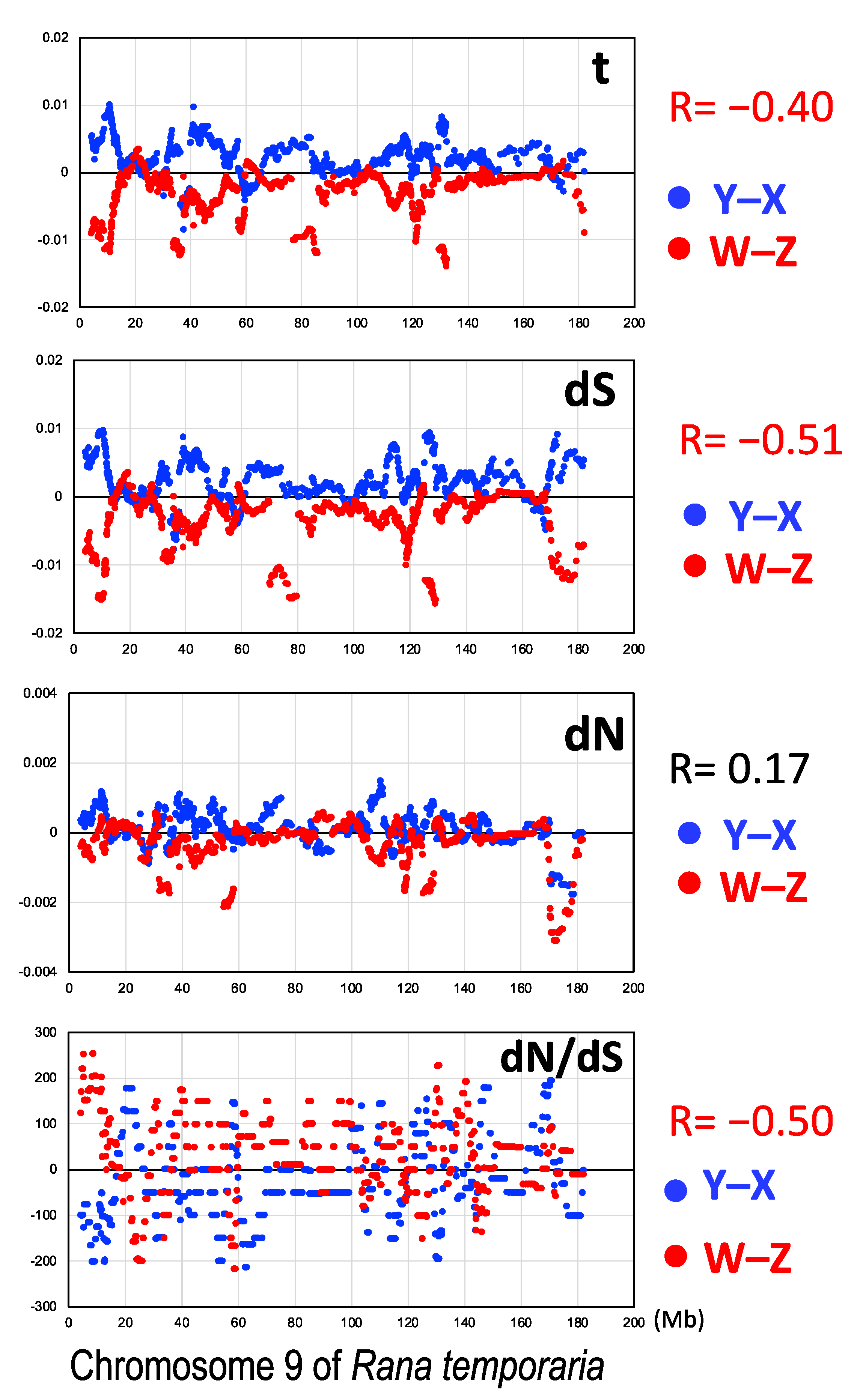
3.3. Male-Biased Mutation
3.4. Female-Biased dN/dS Ratio
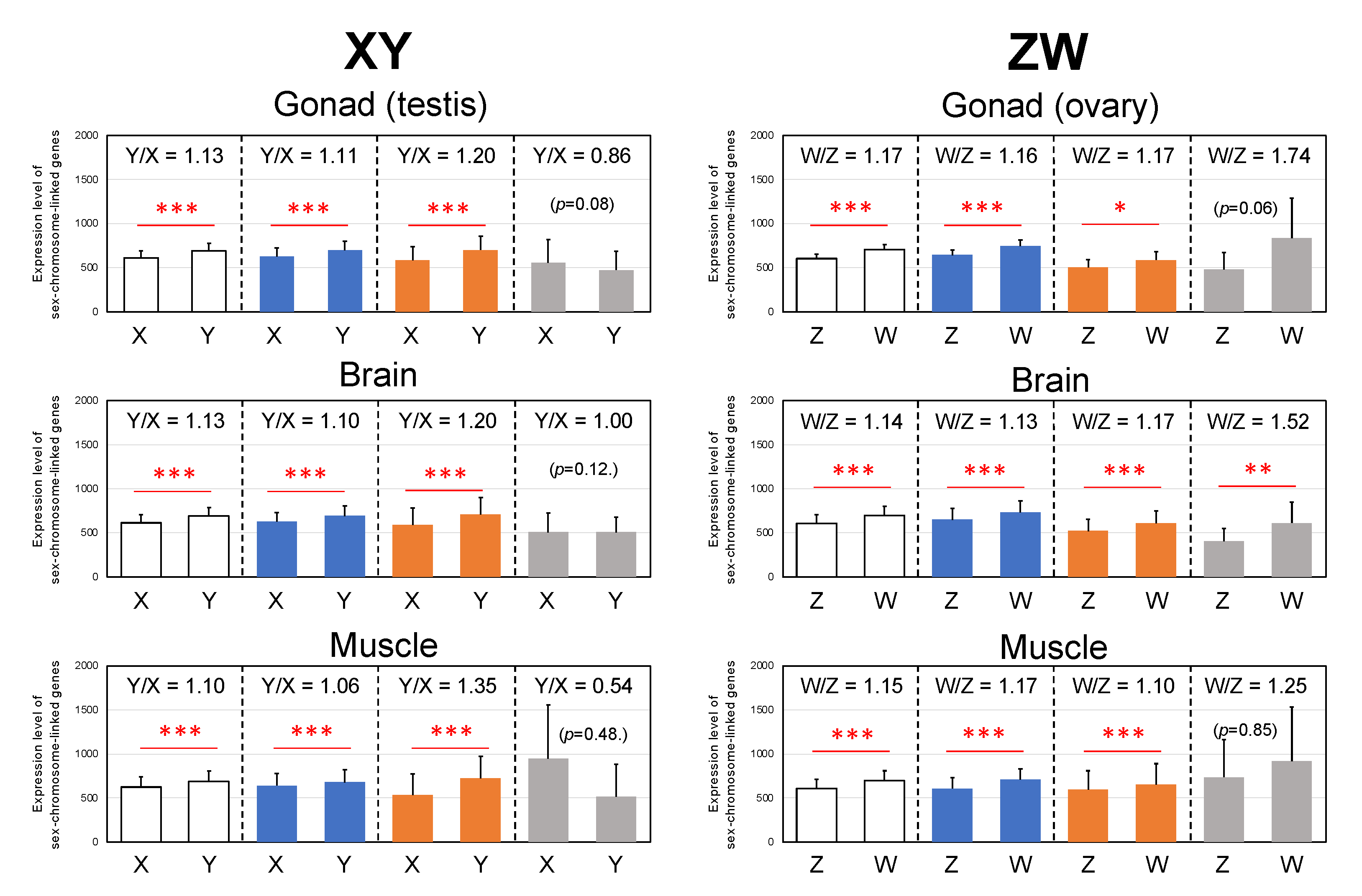
3.5. Y- and W-Biased Expressions
4. Discussion
4.1. Three Evolutionary Strata of the Sex Chromosomes in G. rugosa
4.2. Male-Driven Mutation
4.3. Constraints by Negative Selection on Sex-Linked Genes
4.4. Y- and W-Biased Expression
4.5. Unique Terminal Region of the Sex Chromosomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bull, J.J. Evolution of Sex Determining Mechanisms; Benjamin/Cummings Pub. Co.: Menlo Park, CA, USA, 1983. [Google Scholar]
- Graves, J.A.M. Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annu. Rev. Genet. 2008, 42, 565–586. [Google Scholar] [CrossRef]
- O’Meally, D.; Ezaz, T.; Georges, A.; Sarre, S.D.; Graves, J.A.M. Are some chromosomes particularly good at sex? Insights from amniotes. Chromosome Res. 2012, 20, 7–19. [Google Scholar] [CrossRef]
- Bachtrog, D.; Mank, J.E.; Peichel, C.L.; Kirkpatrick, M.; Otto, S.P.; Ashman, T.L.; Hahn, M.W.; Kitano, J.; Mayrose, I.; Ming, R.; et al. Sex determination: Why so many ways of doing it? PLoS Biol. 2014, 12, e1001899. [Google Scholar] [CrossRef] [PubMed]
- Miura, I. An evolutionary witness: The frog rana rugosa underwent change of heterogametic sex from XY male to ZW female. Sex Dev. 2007, 1, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, D.L.; Lavanchy, G.; Sermier, R.; Sredl, M.J.; Miura, I.; Borzée, A.; Barrow, L.N.; Canestrelli, D.; Crochet, P.A.; Dufresnes, C.; et al. A rapid rate of sex-chromosome turnover and non-random transitions in true frogs. Nat. Commun. 2018, 9, 4088. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Hamaguchi, S. Novel sex-determining genes in fish and sex chromosome evolution. Dev. Dyn. 2013, 242, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, T.; Shams, F.; Yanagi, H.; Ohnishi, T.; Toda, M.; Lin, S.M.; Mawaribuchi, S.; Shimizu, N.; Ezaz, T.; Miura, I. Highly rapid and diverse sex chromosome evolution in the Odorrana frog species complex. Dev. Growth Differ. 2022, 64, 279–289. [Google Scholar] [CrossRef]
- Agulnik, A.I.; Bishop, C.E.; Lerner, J.L.; Agulnik, S.I.; Solovyev, V.V. Analysis of mutation rates in the SMCY/SMCX genes shows that mammalian evolution is male driven. Mamm. Genome 1997, 8, 134–138. [Google Scholar] [CrossRef]
- Wyckoff, G.J.; Li, J.; Wu, C.I. Molecular evolution of functional genes on the mammalian Y chromosome. Mol. Biol. Evol. 2002, 19, 1633–1636. [Google Scholar] [CrossRef]
- Yi, S.; Charlesworth, B. Contrasting patterns of molecular evolution of the genes on the new and old sex chromosomes of Drosophila miranda. Mol. Biol. Evol. 2000, 17, 703–717. [Google Scholar] [CrossRef]
- Bachtrog, D. Evidence for male-driven evolution in Drosophila. Mol. Biol. Evol. 2008, 25, 617–619. [Google Scholar] [CrossRef]
- Filatov, D.A.; Charlesworth, D. Substitution rates in the X- and Y-linked genes of the plants, Silene latifolia and S. dioica. Mol. Biol. Evol. 2002, 19, 898–907. [Google Scholar] [CrossRef][Green Version]
- Nicolas, M.; Marais, G.; Hykelova, V.; Janousek, B.; Laporte, V.; Vyskot, B.; Mouchiroud, D.; Negrutiu, I.; Charlesworth, D.; Monéger, F. A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol. 2005, 3, e4. [Google Scholar] [CrossRef]
- Berlin, S.; Ellegren, H. Fast accumulaton of nonsynonymous mutations on the female-specific W chromosome in birds. J. Mol. Evol. 2006, 62, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.E. Male and female heterogamety in populations of Chironomus tentans (Diptera: Chironomidae). Canad. Entomol. 1971, 103, 369–372. [Google Scholar] [CrossRef]
- Nishioka, M.; Miura, I.; Saitoh, K. Sex chromosomes of Rana rugosa with special reference to local differences in sex determining mechanism. Sci. Rep. Lab. Amphib. Biol. Hiroshima Univ. 1993, 12, 55–81. [Google Scholar]
- Nishioka, M.; Hanada, H.; Miura, I.; Ryuzaki, M. Four kinds of sex chromosomes in Rana rugosa. Sci. Rep. Lab. Amphib. Biol. Hiroshima Univ. 1994, 13, 1–34. [Google Scholar]
- Ogata, M.; Lee, J.Y.; Kim, S.; Ohtani, H.; Sekiya, K.; Igarashi, T.; Hasegawa, Y.; Ichikawa, Y.; Miura, I. The prototype of sex chromosomes found in Korean populations of Rana rugosa. Cytogenet. Genome Res. 2002, 99, 185–193. [Google Scholar] [CrossRef]
- Ogata, M.; Hasegawa, Y.; Ohtani, H.; Mineyama, M.; Miura, I. The ZZ/ZW sex-determining mechanism originated twice and independently during evolution of the frog, Rana rugosa. Heredity 2008, 100, 92–99. [Google Scholar] [CrossRef]
- Ogata, M.; Suzuki, K.; Yuasa, Y.; Miura, I. Sex chromosome evolution from a heteromorphic to a homomorphic system by inter-population hybridization in a frog. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200105. [Google Scholar] [CrossRef]
- Shimada, T.; Matsui, M.; Ogata, M.; Miura, I.; Tange, M.; Min, M.S.; Eto, K. Genetic and morphological variation analyses of Glandirana rugosa with description of a new species (Anura, Ranidae). Zootaxa 2022, 5174, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Miura, I.; Ohtani, H.; Nakamura, M.; Ichikawa, Y.; Saitoh, K. The origin and differentiation of the heteromorphic sex chromosomes Z, W, X, and Y in the frog Rana rugosa, inferred from the sequences of a sex-linked gene, ADP/ATP translocase. Mol. Biol. Evol. 1998, 15, 1602–1609. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogata, M.; Ohtani, H.; Igarashi, T.; Hasegawa, Y.; Ichikawa, Y.; Miura, I. Change of the heterogametic sex from male to female in the frog. Genetics 2003, 164, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Mawaribuchi, S.; Ito, M.; Ogata, M.; Oota, H.; Katsumura, T.; Takamatsu, N.; Miura, I. Meiotic recombination counteracts male-biased mutation (male-driven evolution). Proc. Biol. Sci. 2016, 283, 20152691. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 18 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Streicher, J.W. Wellcome Sanger Institute Tree of Life programme; Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective; Tree of Life Core Informatics collective; Darwin Tree of Life Consortium. The genome sequence of the common frog, Rana temporaria Linnaeus 1758. Wellcome Open Res. 2021, 6, 286. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Nariai, N.; Kojima, K.; Mimori, T.; Kawai, Y.; Nagasaki, M. A Bayesian approach for estimating allele-specific expression from RNA-Seq data with diploid genomes. BMC Genom. 2016, 17 (Suppl. 1), 2. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Miura, I.; Hanada, H.; Ichikawa, Y. Alteration of the sex determining system resulting from structural change of the sex chromosomes in the frog Rana rugosa. J. Exp. Zool. 2000, 286, 313–319. [Google Scholar] [CrossRef]
- Miura, I.; Ezaz, T.; Ohtani, H.; Graves, J.A.M. Molecular characterization of ZW sex chromosomes and the prototype chromosome in the frog Rana rugosa. Adv. Chromosome Sci. 2009, 3, 101. [Google Scholar]
- Molescalchi, A. Amphibia. In Cytotaxonomy and Vertebrate Evolution; Chiarelli, A.B., Capanna, E., Eds.; Academic Press: London, UK; New York, NY, USA, 1973; pp. 233–348. [Google Scholar]
- Okumoto, H. Studies on meioses in male hybrids and triploids in the Rana nigromaculata group. Sci. Rep. Lab. Amphibian Biol. Hiroshima Univ. 1980, 4, 201–216. [Google Scholar]
- Ohtani, H. Lampbrush chromosomes of Rana nigromaculata, R. brevipoda, R. plancyi chosenica, R. p.fukienensis and their reciprocal hybrids. Sci. Rep. Lab. Amphibian Biol. Hiroshima Univ. 1990, 10, 165–221. [Google Scholar]
- Ogata, M.; Shams, S.; Yoshimura, Y.; Ezaz, T.; Miura, I. W Chromosome Evolution by Repeated Recycling in the Frog Glandirana rugosa. DNA 2022, 2, 172–184. [Google Scholar] [CrossRef]
- Ogata, M.; Lambert, M.; Ezaz, T.; Miura, I. Reconstruction of female heterogamety from admixture of XX-XY and ZZ-ZW sex chromosome systems within a frog species. Mol. Ecol. 2018, 27, 4078–4089. [Google Scholar] [CrossRef]
- Pandey, R.S.; Sayres, M.A.W.; Azad, R.K. Detecting evolutionary strata on the human x chromosome in the absence of gametologous y-linked sequences. Genome Biol. Evol. 2013, 5, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Bellott, D.W.; Skaletsky, H.; Cho, T.J.; Brown, L.; Locke, D.; Chen, N.; Galkina, S.; Pyntikova, T.; Koutseva, N.; Graves, T.; et al. Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nat. Genet. 2017, 49, 387–394. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Xu, X.; Witt, C.; Deng, Y.; Chen, G.; Meng, G.; Feng, S.; Xu, L.; Szekely, T.; et al. Phylogeny and sex chromosome evolution of Palaeognathae. J. Genet. Genom. 2022, 49, 109–119. [Google Scholar] [CrossRef]
- Miyata, T.; Hayashida, H.; Kuma, K.; Mitsuyasu, K.; Yasunaga, T. Male-driven molecular evolution: A model and nucleotide sequence analysis. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 1987; Volume 52, pp. 863–867. [Google Scholar]
- Shimmin, L.C.; Chang, B.H.; Li, W.H. Male-driven evolution of DNA sequences. Nature 1993, 362, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Fridolfsson, A.K. Male-driven evolution of DNA sequences in birds. Nat. Genet. 1997, 17, 182–184. [Google Scholar] [CrossRef]
- Vogel, F.; Motulsky, A.G. Human Genetics: Problems and Approaches; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Zhu, Z.X.; Matsubara, K.; Shams, F.; Dobry, J.; Wapstra, E.; Gamble, T.; Sarre, S.D.; Georges, A.; Graves, J.A.M.; Zhou, Q.; et al. Diversity of reptile sex chromosome evolution revealed by cytogenetic and linked-read sequencing. Zool. Res. 2022, 43, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Bachtrog, D. Adaptation shapes patterns of genome evolution on sexual and asexual chromosomes in Drosophila. Nat. Genet. 2003, 34, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, D.T.; Filatov, D.A. Positive and negative selection on mammalian Y chromosomes. Mol. Biol. Evol. 2005, 22, 1423–1432. [Google Scholar] [CrossRef]
- Miura, I.; Ohtani, H.; Kashiwagi, A.; Hanada, H.; Nakamura, M. Structural differences between XX and ZW sex lampbrush chromosomes in Rana rugosa females (Anura: Ranidae). Chromosoma 1996, 105, 237–241. [Google Scholar] [CrossRef]
- Miura, I.; Ohtani, H.; Ogata, M. Independent degeneration of the W and Y sex chromosomes in frog Rana rugosa. Chromosome Res. 2012, 20, 47–55. [Google Scholar] [CrossRef]
- Dixon, G.; Kitano, J.; Kirkpatrick, M. The Origin of a New Sex Chromosome by Introgression between Two Stickleback Fishes. Mol. Biol. Evol. 2019, 36, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.K.; Naqvi, S.; Chmátal, L.; Chick, J.M.; Mitchell, R.N.; Gygi, S.P.; Skaletsky, H.; Page, D.C. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 2020, 30, 860–873. [Google Scholar] [CrossRef]
- Lenormand, T.; Fyon, F.; Sun, E.; Roze, D. Sex Chromosome Degeneration by Regulatory Evolution. Curr. Biol. 2020, 30, 3001–3006.e5. [Google Scholar] [CrossRef] [PubMed]
- Ayers, K.L.; Davidson, N.M.; Demiyah, D.; Roeszler, K.N.; Grützner, F.; Sinclair, A.H.; Oshlack, A.; Smith, C.A. RNA sequencing reveals sexually dimorphic gene expression before gonadal differentiation in chicken and allows comprehensive annotation of the W-chromosome. Genome Biol. 2013, 14, R26. [Google Scholar] [CrossRef] [PubMed]
- Pál, C.; Papp, B.; Hurst, L.D. Highly expressed genes in yeast evolve slowly. Genetics 2001, 158, 927–931. [Google Scholar] [CrossRef]
- Drummond, D.A.; Bloom, J.D.; Adami, C.; Wilke, C.O.; Arnold, F.H. Why highly expressed proteins evolve slowly. Proc. Natl. Acad. Sci. USA 2005, 102, 14338–14343. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Nakamoto, M.; Morishima, K.; Yamashita, R.; Yamashita, T.; Sasaki, K.; Kuruma, Y.; Mizuno, N.; Suzuki, M.; Okada, Y.; et al. A SNP in a Steroidogenic Enzyme Is Associated with Phenotypic Sex in Seriola Fishes. Curr. Biol. 2019, 29, 1901–1909.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Yuan, J.; Liu, F.; Hong, X.; Yu, L.; Chen, C.; Li, W.; Ni, W.; Liu, H.; et al. Chromosome-level genome assembly of Asian yellow pond turtle (Mauremys mutica) with temperature-dependent sex determination system. Sci. Rep. 2022, 12, 7905. [Google Scholar] [CrossRef]
- Bione, S.; Rizzolio, F.; Sala, C.; Ricotti, R.; Goegan, M.; Manzini, M.C.; Battaglia, R.; Marozzi, A.; Vegetti, W.; Dalprà, L.; et al. Mutation analysis of two candidate genes for premature ovarian failure, DACH2 and POF1B. Hum. Reprod. 2004, 19, 2759–2766. [Google Scholar] [CrossRef]
- Fortriede, J.D.; Pells, T.J.; Chu, S.; Chaturvedi, P.; Wang, D.; Fisher, M.E.; James-Zorn, C.; Wang, Y.; Nenni, M.J.; Burns, K.A.; et al. Xenbase: Deep integration of GEO & SRA RNA-seq and ChIP-seq data in a model organism database. Nucleic Acids Res. 2020, 48, D776–D782. [Google Scholar] [CrossRef]
- Miura, I.; Shams, F.; Jeffries, D.L.; Katsura, Y.; Mawaribuchi, S.; Perrin, N.; Ito, M.; Ogata, M.; Ezaz, T. Identification of ancestral sex chromosomes in the frog Glandirana rugosa bearing XX-XY and ZZ-ZW sex-determining systems. Mol. Ecol. 2022, 31, 3859–3870. [Google Scholar] [CrossRef]
- Miura, I. Sex Determination and Sex Chromosomes in Amphibia. Sex Dev. 2017, 11, 298–306. [Google Scholar] [CrossRef]
- Uno, Y.; Nishida, C.; Yoshimoto, S.; Ito, M.; Oshima, Y.; Yokoyama, S.; Nakamura, M.; Matsuda, Y. Diversity in the origins of sex chromosomes in anurans inferred from comparative mapping of sexual differentiation genes for three species of the Raninae and Xenopodinae. Chromosome Res. 2008, 16, 999–1011. [Google Scholar] [CrossRef]
- Oshima, Y.; Uno, Y.; Matsuda, Y.; Kobayashi, T.; Nakamura, M. Molecular cloning and gene expression of Foxl2 in the frog Rana rugosa. Gen. Comp. Endocrinol. 2008, 159, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Suda, M.; Sakamoto, D.; Iwasaki, T.; Matsuo, Y.; Uno, Y.; Matsuda, Y.; Nakamura, Y.; Maekawa, S.; Katsu, Y.; et al. Molecular cloning and characterization of anti-Müllerian hormone (AMH) from the Japanese wrinkled frog, Rana rugosa. Endocrinology 2015, 156, 1914–1923. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Sex Chromosome | N (1) | S (2) | No. of dN (3) | No. of dS (4) | dN (5) | dS (6) | dN/dS |
|---|---|---|---|---|---|---|---|---|
| All | X | 866,897.7 | 316,932.3 | 646.1 * | 1471.4 * | 0.0007 | 0.0046 | 0.1605 |
| Y | 866,897.7 | 316,932.3 | 869.1 * | 2304.7 * | 0.0010 | 0.0073 | 0.1379 | |
| Z | 866,897.7 | 316,932.3 | 754.5 *** | 1976.4 *** | 0.0009 | 0.0062 | 0.1396 | |
| W | 866,897.7 | 316,932.3 | 571.2 *** | 1155.1 *** | 0.0007 | 0.0036 | 0.1808 | |
| XW/YZ | X | 651,460.7 | 239,629.3 | 333.8 | 808.0 | 0.0005 | 0.0034 | 0.1520 |
| Y | 651,460.7 | 239,629.3 | 548.8 | 1430.6 | 0.0008 | 0.0060 | 0.1411 | |
| Z | 651,460.7 | 239,629.3 | 469.6 * | 1230.9 * | 0.0007 | 0.0051 | 0.1403 | |
| W | 651,460.7 | 239,629.3 | 323.1 * | 706.6 * | 0.0005 | 0.0029 | 0.1682 | |
| XY/ZW | X | 199,657.2 | 71,668.8 | 265.6 * | 575.5 * | 0.0013 | 0.0080 | 0.1657 |
| Y | 199,657.2 | 71,668.8 | 278.5 * | 785.2 * | 0.0014 | 0.0110 | 0.1273 | |
| Z | 199,657.2 | 71,668.8 | 240.2 ** | 659.2 ** | 0.0012 | 0.0092 | 0.1308 | |
| W | 199,657.2 | 71,668.8 | 216.6 ** | 417.1 ** | 0.0011 | 0.0058 | 0.1864 | |
| XZ/YW | X | 15,779.8 | 5634.2 | 46.7 | 87.9 | 0.0030 | 0.0156 | 0.1897 |
| Y | 15,779.8 | 5634.2 | 41.8 | 88.9 | 0.0026 | 0.0158 | 0.1679 | |
| Z | 15,779.8 | 5634.2 | 44.7 * | 86.3 * | 0.0028 | 0.0153 | 0.1849 | |
| W | 15,779.8 | 5634.2 | 31.5 * | 31.4 * | 0.0020 | 0.0056 | 0.3582 |
| Expression (TPM) | Ratio | Expression (TPM) | Ratio | Chromosomal | |||||
|---|---|---|---|---|---|---|---|---|---|
| ID/Gene Name | Cluster | Tissue | Y | X | Y/X | Z | W | W/Z | Position (bp) (1) |
| 120914319 | XY/ZW | gonad | 270.5 | 96.2 | 2.81 | 252.5 | 252.5 | 1.00 | 168935130 |
| HSD17B8 | brain | 130.5 | 25.3 | 5.16 | 66.7 | 66.7 | 1.00 | ||
| muscle | 158.6 | 61.3 | 2.59 | 49.4 | 49.4 | 1.00 | |||
| 120913254 | XY/ZW | gonad | 2264.9 | 0.3 | 7088.2 | 187.0 | 1956.9 | 10.5 | 31494656 |
| NELFE | brain | 1912.9 | 0.6 | 3112.5 | 122.7 | 1486.4 | 12.1 | ||
| muscle | 737.3 | 0.7 | 1066.1 | 63.1 | 757.2 | 12.0 | |||
| 120914576 | XY/ZW | gonad | 299.5 | 37.5 | 7.98 | 3.3 | 35.2 | 10.7 | 109121688 |
| LOC120914576 | brain | 239.9 | 24.1 | 9.96 | 34.6 | 223.0 | 6.4 | ||
| E3 SUMO-protein ligaseZBED1-like | muscle | 226.7 | 10.6 | 21.44 | 31.9 | 116.5 | 3.6 | ||
| 120914364 | XY/ZW | gonad | 58.6 | 63.2 | 0.93 | 1.1 | 371.8 | 332.9 | 178509991 |
| Dach2 | brain | 500.2 | 231.2 | 2.16 | 0.8 | 576.5 | 707.4 | ||
| muscle | 52.1 | 18.4 | 2.83 | 2.0 | 103.2 | 50.4 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mawaribuchi, S.; Ito, M.; Ogata, M.; Yoshimura, Y.; Miura, I. Parallel Evolution of Sex-Linked Genes across XX/XY and ZZ/ZW Sex Chromosome Systems in the Frog Glandirana rugosa. Genes 2023, 14, 257. https://doi.org/10.3390/genes14020257
Mawaribuchi S, Ito M, Ogata M, Yoshimura Y, Miura I. Parallel Evolution of Sex-Linked Genes across XX/XY and ZZ/ZW Sex Chromosome Systems in the Frog Glandirana rugosa. Genes. 2023; 14(2):257. https://doi.org/10.3390/genes14020257
Chicago/Turabian StyleMawaribuchi, Shuuji, Michihiko Ito, Mitsuaki Ogata, Yuri Yoshimura, and Ikuo Miura. 2023. "Parallel Evolution of Sex-Linked Genes across XX/XY and ZZ/ZW Sex Chromosome Systems in the Frog Glandirana rugosa" Genes 14, no. 2: 257. https://doi.org/10.3390/genes14020257
APA StyleMawaribuchi, S., Ito, M., Ogata, M., Yoshimura, Y., & Miura, I. (2023). Parallel Evolution of Sex-Linked Genes across XX/XY and ZZ/ZW Sex Chromosome Systems in the Frog Glandirana rugosa. Genes, 14(2), 257. https://doi.org/10.3390/genes14020257