
Identification of a Small Supernumerary Marker Chromosome in a Turner Syndrome Patient with Karyotype mos 46,X,+mar/45,X

 ,
,
Abstract
1. Introduction
2. Materials and Method
2.1. Clinical Study
2.2. Karyotype and FISH
2.3. Microarray Experiment
3. Results
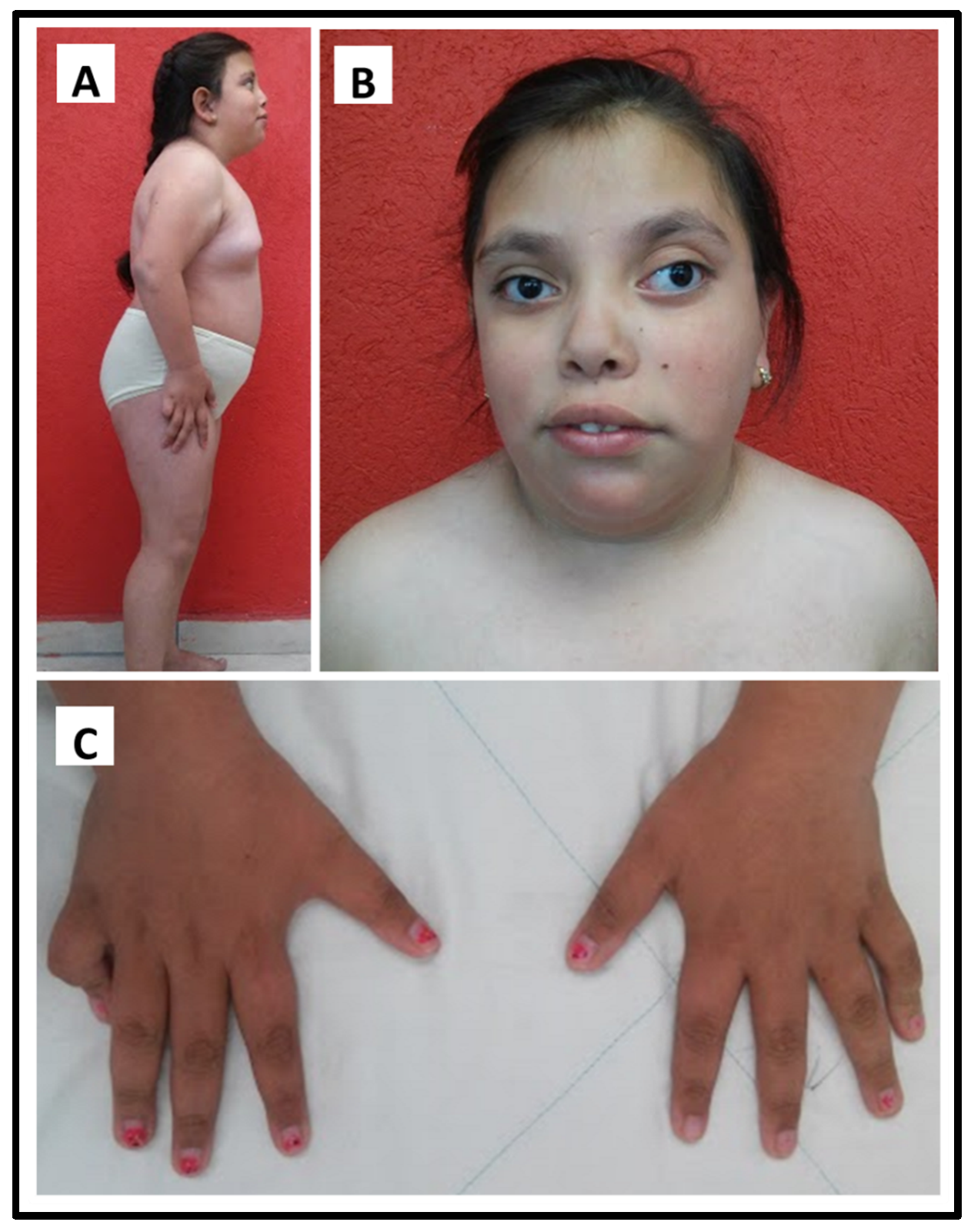
3.1. Clinical Study
3.2. Cytogenetic Study
4. Discussion

{kind=link}
{kind=link}
{kind=link}
| Karyotype | Cells with 45,X (%) | Short Stature | Ovarian Failure | ID/DD | XIST Present | Unusual Findings for TS | Reference |
|---|---|---|---|---|---|---|---|
| mos 46,X,+mar(X)(p11.21q21.1)/45,X | 20 | + | + | + | + | Facial dysmorphism Divergent strabismusLimited extension of both elbows | Present case |
| mos 45,X/46,X,r(X) | 88 | + | + | + | N.A. | Facial dysmorphism. Divergent strabismusLimb contractures Syndactyly | Kushnick (1987) [20] |
| mos 45,X/46,X,r(X) | 42 | + | N.A. | + | N.A. | Facial dysmorphism Ventricular septal defect Foramen ovale Patent ductus arteriosus | Lindgren (1992) [21] |
| mos 45,X/46,X,r(X) | 82 | + | N.A. | + | N.A | Facial dysmorphism | Cole (1994) [22] |
| mos 45,X/46,X,r(X)(p11.23q11.2)/ 46,X,dic(X)(p11) | 82 | + | + | − | − | Asymmetry of the left arm | Robson (1994) [23] |
| mos 45,X/46,X,r(X)(p21q22)/ 46,X,min(X)(p11.1q11.1) | 40 | + | N.A. | + | + − | Hyper flexible joints | Wolff (1994) [26] |
| mos 45,X/46,X,r(X)/ 47,X,r1(X),+r2(X) | N.A. | + | N.A. | + | − + | Strabismus Scoliosis | Cantú (1995) [27] |
| mos 45,X/46,X,del(X)(q21.3-qter)/46X,r(X)(p22.3q13.2) | 80 | + | + | + | + | Cranial and facial asymmetry with dysmorphism Contractures of the elbow Seizures Maternal iso UPD | Migeon (1996) [28] |
| mos 45,X/46,X,r(X)(p21.2q13.2) | 83 | + | + | + | − | Facial dysmorphism | Turner (2000) [24] |
| mos 45,X/r(X)(p11.22q11.2) | 12 | + | N.A. | N.A. | − | Kabuki-like facies | Turner (2000) [24] |
| mos 45,X/r(X)(p11q11) | 70 | + | + | − | − | Kabuki-like facies | Turner (2000) [24] |
| mos 45,X/46,X,r(X)(p11q21.1) | 13 | + | N.A. | + | + | Facial dysmorphism Microcephaly | Kubota (2002) [32] |
| mos 45,X/46,X,r(X)(p11q21.1) | 10 | + | N.A. | + | + | Facial dysmorphism Brachydactyly | Kubota (2002) [32] |
| mos 45,X/46,X,r(X)(p11.3q13) | 28 | + | N.A. | + | + | Facial dysmorphism Syndactyly Brachydactyly | Tomkins (2002) [29] |
| mos 45,X/r(X)(p11q11) | 10 | + | − | − | − | Alopecia universalis | BouayedAbdelmoula (2004) [25] |
| r(X)(p11.3~11.4q13.3)[19%]/ r(X;X)(p11.3~11.4q13.3::p11.3~11.4q13.3)[2%] | 79 | + | N.A. | + | + | N.A | Liehr (2007) [7] |
| r(X)(p11.3q12) aCGH 44.19–64.59 MB | 80 | + | N.A. | + | + | N.A | Liehr (2007) [7] |
| mos 45,X/46,X+mar(X)(q11.11q13.2) | 95 | + | N.A. | + | + | Facial dysmorphism | Kalkan (2016) [30] |
| mos 45,X/46,X,r(X)(p22.11q21.32) | 50 | + | + | − | + | − | Chauhan (2016) |
| mos 45,X/46,X,r(X)(p11.23q21.1) | 57 | + | N.A. | − | + | N.A. | Li (2020) [31] |
| r(X)(p11.22q13.1) aCGH 54.09–67.73 MB | 43 | N.A. | + | + | − | N.A. | Liehr (2023) [9] |
| r(X)(p11.2?2q21~22) | 66 | + | + | + | N.A. | N.A. | Liehr (2023) [9] |
| r(X)(p11.22q22.3) | 43 | + | + | + | + | N.A. | Liehr (2023) [9] |
| r(X)(p11.21q13) | 36 | + | + | + | + | N.A. | Liehr (2023) [9] |
| r(X)(p1?1.2q13.?3) | 77 | + | + | + | + | N.A. | Liehr (2023) [9] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Viuff, M.; Just, J.; Sandahl, K.; Brun, S.; van der Velden, J.; Andersen, N.H.; Skakkebaek, A. The Changing Face of Turner Syndrome. Endocr. Rev. 2022, 44, 33–69. [Google Scholar] [CrossRef] [PubMed]
- Lebenthal, Y.; Levy, S.; Sofrin-Drucker, E.; Nagelberg, N.; Weintrob, N.; Shalitin, S.; De Vries, L.; Tenenbaum, A.; Phillip, M.; Lazar, L. The natural history of metabolic comorbidities in Turner syndrome from childhood to early adulthood: Comparison between 45, X monosomy and other karyotypes. Front. Endocrinol 2018, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Zelinska, N.; Shevchenko, I.; Globa, E. Nationwide study of turner syndrome in Ukrainian children: Prevalence, genetic variants and phenotypic features. JCRPE J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Frelich, J.; Lepska, K.; Jeż, W.; Irzyniec, T.; Lepska, K.; Jeż, W. New insights into clinical features, karyotypes, and age at diagnosis in women with Turner syndrome. Endokrynol. Pol. 2019, 70, 342–349. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24002961 (accessed on 20 November 2022). [CrossRef] [PubMed]
- Noordman, I.; Duijnhouwer, A.; Kapusta, L.; Kempers, M.; Roeleveld, N.; Schokking, M.; Smeets, D.; Freriks, K.; Timmers, H.; van Alfen-van der Velden, J. Phenotype in girls and women with Turner syndrome: Association between dysmorphic features, karyotype and cardio-aortic malformations. Eur. J. Med. Genet. 2018, 61, 301–306. [Google Scholar] [CrossRef]
- Liehr, T.; Mrasek, K.; Hinreiner, S.; Reich, D.; Ewers, E.; Bartels, I.; Seidel, J.; Emmanuil, N.; Petesen, M.; Polityko, A.; et al. Small supernumerary marker chromosomes (sSMC) in patients with a 45, X/46, X, +mar karyotype—17 New cases and a review of the literature. Sex. Dev. 2007, 1, 353–362. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, Z.; Xia, P.; Lai, Y.; Wei, Y.; Liu, Y.; Chen, J.; Qin, L.; Xie, L.; Wang, H. Clinical manifestation and cytogenetic analysis of 607 patients with Turner syndrome. Chin. J. Med. Genet. 2017, 34, 61–64. Available online: https://pubmed.ncbi.nlm.nih.gov/28186596/ (accessed on 29 November 2022).
- Liehr, T. Small Supernumerary Marker Chromosomes. Available online: http://cs-tl.de/DB/CA/sSMC/0-Start.html (accessed on 2 January 2022).
- Chauhan, P.; Jaiswal, S.K.; Lakhotia, A.R.; Rai, A.K. Molecular cytogenetic characterization of two Turner syndrome patients with mosaic ring X chromosome. J. Assist. Reprod Genet. 2016, 33, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.; Chae, H.; Kim, J.; Son, J.O.; Kim, S.C.; Koo, B.K.; Kim, M.; Kim, Y.; Park, I.Y.; Sung, I.K. Identification of small marker chromosomes using microarray comparative genomic hybridization and multicolor fluorescent in situ hybridization. Mol. Cytogenet 2016, 9, 61. [Google Scholar] [CrossRef]
- Lawce, H.J.; Brown, M.G. Cytogenetics: An overview. In The AGT Cytogenetics Laboratory Manual; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 25–85. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN—An International System for Human Cytogenomic Nomenclature; Karger Publishers: Basel, Switzerland, 2020. [Google Scholar]
- Siniscalchi, C.; di Palo, A.; Russo, A.; Potenza, N. The lncRNAs at X Chromosome Inactivation Center: Not Just a Matter of Sex Dosage Compensation. Int. J. Mol. Sci. 2022, 23, 611. [Google Scholar] [CrossRef] [PubMed]
- Sierra, I.; Anguera, M.C. Enjoy the silence: X-chromosome inactivation diversity in somatic cells. Curr. Opin. Genet. Dev. 2019, 55, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R. Choosing the Active X: The Human Version of X Inactivation. Trends Genet. 2017, 33, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fan, J.; Wang, Y. X-Chromosome Inactivation and Related Diseases. Genet. Res. 2022, 2022, 1391807. [Google Scholar] [CrossRef] [PubMed]
- Leppig, K.A.; Disteche, C.M. Ring X and other structural X chromosome abnormalities: X inactivation and phenotype. Semin. Reprod. Med. 2001, 19, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Jani, M.M.; Torchia, B.S.; Shashidhar Pai, G.; Migeon, B.R. Molecular characterization of tiny ring X chromosomes from females with functional X chromosome disomy and lack of cis X inactivation. Genomics 1995, 27, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kushnick, T.; Irons, T.G.; Wiley, J.E.; Gettig, E.A.; Rao, K.W.; Bowyer, S.; Opitz, J.M.; Reynolds, J.F. 45X/46X, r(X) with Syndactyly and Severe Mental Retardation. Am. J. Med. Genet. 1987, 28, 567–574. [Google Scholar] [CrossRef]
- Lindgren, V.; Chen, C.P.; Bryke, C.R.; Lichter, P.; Page, D.C.; Yang-Feng, T.L. Cytogenetic and molecular characterization of marker chromosomes in patients with mosaic 45, X karyotypes. Hum. Genet. 1992, 88, 393–398. [Google Scholar] [CrossRef]
- Cole, H.; Huang, B.; Salbert, B.A.; Brown, J.; Howard-Peebles, P.N.; Black, S.H.; Dorfmann, A.; Febles, O.R.; Stevens, C.A.; Jackson-Cook, C. Mental Retardation and Ullrich-Turner Syndrome in Cases With 45, W46, X, + mar: Additional Support for the Loss of the X-Inactivation Center Hypothesis. Am. J. Med. Genet. 1994, 52, 136–145. [Google Scholar] [CrossRef]
- Robson, L.; Jackson, J.; Cowell, C.; Sillence, D.; Smith, A. Novel karyotype in the Ullrich-Turner syndrome—45, X/46, X,r(X)/46, X, dic(X)—Investigated with fluorescence in situ hybridization. Am. J. Med. Genet. 1994, 50, 251–254. [Google Scholar] [CrossRef]
- Turner, C.; Dennis, N.R.; Skuse, D.H.; Jacobs, P.A. Seven ring (X) chromosomes lacking the XIST locus, six with an unexpectedly mild phenotype. Hum. Genet. 2000, 106, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoula, N.B.; Portnoi, M.F.; Amouri, A.; Arladan, A.; Chakroun, M.; Saad, A.; Hchicha, M.; Turki, H.; Rebai, T. Turner syndrome female with a small ring X chromosome lacking the XIST, an unexpectedly mild phenotype and an atypical association with alopecia universalis. Ann. Genet. 2004, 47, 305–313. [Google Scholar] [CrossRef]
- Wolff, D.J.; Brown, C.J.; Schwartz, S.; Duncan, A.M.; Surti, U.; Willard, H.F. Small marker X chromosomes lack the X inactivation center: Implications for karyotype/phenotype correlations. Am. J. Hum. Genet. 1994, 55, 87–95. [Google Scholar]
- Cantu, E.S.; Jacobs, D.F.; Pai, G.S. An atypical Turner syndrome patient with ring X chromosome mosaicism. Ann. Clin. Lab. Sci. 1995, 25, 60–65. [Google Scholar] [PubMed]
- Migeon, B.R.; Jeppesen, P.; Torchia, B.S.; Fu, S.; Dunn, M.A.; Axelman, J.; Schmeckpeper, B.J.; Fantes, J.; Zori, R.T.; Driscoll, D.J. Lack of X inactivation associated with maternal X isodisomy: Evidence for a counting mechanism prior to X inactivation during human embryogenesis. Am. J. Hum. Genet. 1996, 58, 161–170. [Google Scholar] [PubMed]
- Tomkins, D.J.; McDonald, H.L.; Farrell, S.A.; Brown, C.J. Lack of expression of XIST from a small ring X chromosome containing the XIST locus in a girl with short stature, facial dysmorphism and developmental delay. Eur. J. Hum. Genet. 2002, 10, 44–51. [Google Scholar] [CrossRef]
- Kalkan, R.; Özdağ, N.; Bundak, R.; Çirakoğlu, A.; Serakinci, N. A unique mosaic Turner syndrome patient with androgen receptor gene derived marker chromosome. Syst. Biol. Reprod. Med. 2016, 62, 77–83. [Google Scholar] [CrossRef]
- Li, T.; Sang, H.; Chu, G.; Zhang, Y.; Qi, M.; Liu, X.; Cui, W.; Zhao, Y. Genotype-phenotype correlation in 75 patients with small supernumerary marker chromosomes. Mol. Cytogenet 2020, 13, 30. [Google Scholar] [CrossRef]
- Kubota, T.; Wakui, K.; Nakamura, T.; Ohashi, H.; Watanabe, Y.; Yoshino, M.; Kida, T.; Okamoto, N.; Matsumura, M.; Muroya, K.; et al. The proportion of cells with functional X disomy is associated with the severity of mental retardation in mosaic ring X Turner syndrome females. Cytogenet. Genome Res. 2002, 99, 276–284. [Google Scholar] [CrossRef]
- Graff, A.; Donadille, B.; Morel, H.; Villy, M.C.; Bourcigaux, N.; Vatier, C.; Borgel, A.; Khodawardi, A.; Siffroi, J.P.; Christin-Maitre, S. Added value of buccal cell FISH analysis in the diagnosis and management of Turner syndrome. Hum. Reprod. 2020, 35, 2391–2398. Available online: https://academic.oup.com/humrep/article/35/10/2391/5894062 (accessed on 20 November 2022).
- Prakash, S.K. The impact of somatic mosaicism on bicuspid aortic valve and aortic dissection in Turner Syndrome. Am. J. Med. Genet. C Semin Med. Genet. 2019, 181, 7–12. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Rodríguez, M.T.A.; Brukman-Jiménez, S.A.; Cuero-Quezada, I.; Corona-Rivera, J.R.; Corona-Rivera, A.; Serafín-Saucedo, G.; Aguirre-Salas, L.M.; Bobadilla-Morales, L. Identification of a Small Supernumerary Marker Chromosome in a Turner Syndrome Patient with Karyotype mos 46,X,+mar/45,X. Genes 2023, 14, 253. https://doi.org/10.3390/genes14020253
González-Rodríguez MTA, Brukman-Jiménez SA, Cuero-Quezada I, Corona-Rivera JR, Corona-Rivera A, Serafín-Saucedo G, Aguirre-Salas LM, Bobadilla-Morales L. Identification of a Small Supernumerary Marker Chromosome in a Turner Syndrome Patient with Karyotype mos 46,X,+mar/45,X. Genes. 2023; 14(2):253. https://doi.org/10.3390/genes14020253
Chicago/Turabian StyleGonzález-Rodríguez, María Teresa Alejandra, Sinhue Alejandro Brukman-Jiménez, Idalid Cuero-Quezada, Jorge Román Corona-Rivera, Alfredo Corona-Rivera, Graciela Serafín-Saucedo, Liuba M. Aguirre-Salas, and Lucina Bobadilla-Morales. 2023. "Identification of a Small Supernumerary Marker Chromosome in a Turner Syndrome Patient with Karyotype mos 46,X,+mar/45,X" Genes 14, no. 2: 253. https://doi.org/10.3390/genes14020253
APA StyleGonzález-Rodríguez, M. T. A., Brukman-Jiménez, S. A., Cuero-Quezada, I., Corona-Rivera, J. R., Corona-Rivera, A., Serafín-Saucedo, G., Aguirre-Salas, L. M., & Bobadilla-Morales, L. (2023). Identification of a Small Supernumerary Marker Chromosome in a Turner Syndrome Patient with Karyotype mos 46,X,+mar/45,X. Genes, 14(2), 253. https://doi.org/10.3390/genes14020253