
Linear Diagnostic Procedure Elicited by Clinical Genetics and Validated by mRNA Analysis in Neuronal Ceroid Lipofuscinosis 7 Associated with a Novel Non-Canonical Splice Site Variant in MFSD8


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Study and Literature Review
2.2. Analyses on Genomic DNA
2.3. Analyses on cDNA
3. Results
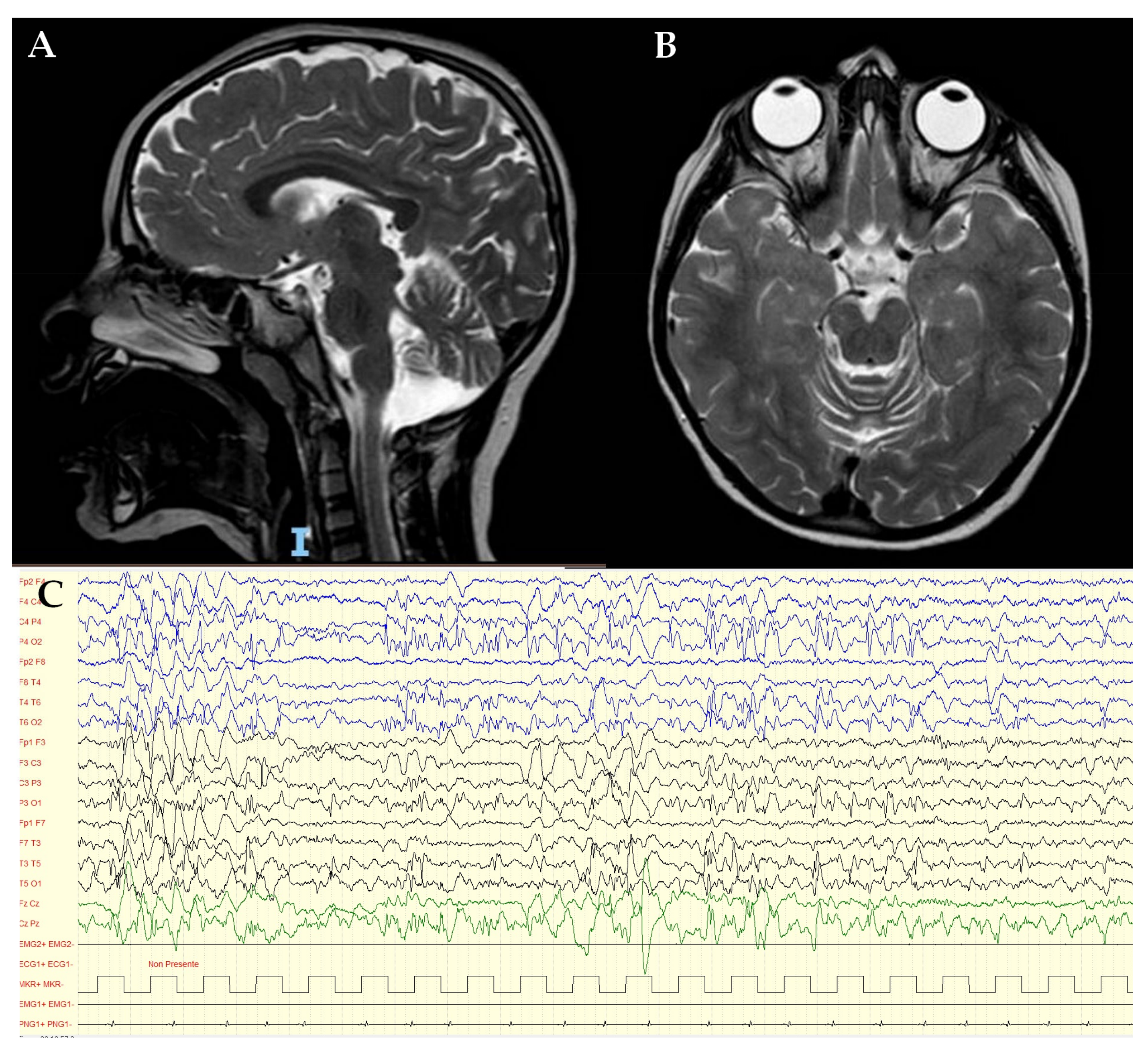
3.1. Clinical Report
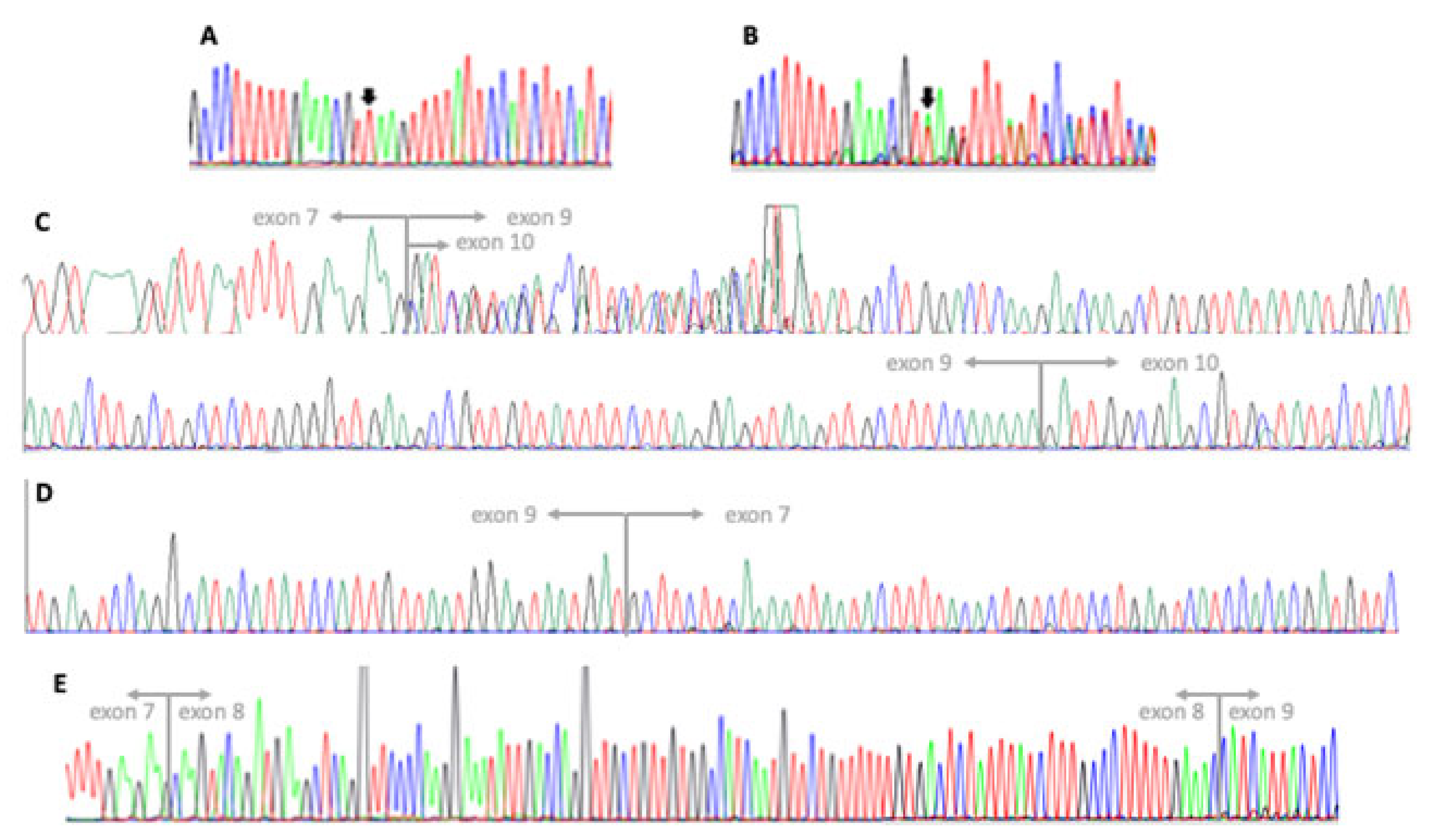
3.2. Genetic Testing
4. Discussion
- (1)
- Null or almost null alleles, leading to a nearly complete loss of protein function;
- (2)
- Hypomorphic alleles, with some degree of residual protein function.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haltia, M.; Goebel, H.H. The Neuronal Ceroid-Lipofuscinoses: A Historical Introduction. Biochim. Biophys. Acta 2013, 1832, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.D.; Tarczyluk, M.A.; Nelvagal, H.R. Towards a New Understanding of NCL Pathogenesis. Biochim. Biophys. Acta 2015, 1852, 2256–2261. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL Neuropathology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 2262–2266. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between Genotype, Ultrastructural Morphology and Clinical Phenotype in the Neuronal Ceroid Lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef]
- Jadav, R.H.; Sinha, S.; Yasha, T.C.; Aravinda, H.; Gayathri, N.; Rao, S.; Bindu, P.S.; Satishchandra, P. Clinical, Electrophysiological, Imaging, and Ultrastructural Description in 68 Patients with Neuronal Ceroid Lipofuscinoses and Its Subtypes. Pediatr. Neurol. 2014, 50, 85–95. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.-Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.-K.; et al. The Novel Neuronal Ceroid Lipofuscinosis Gene MFSD8 Encodes a Putative Lysosomal Transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef]
- Sharifi, A.; Kousi, M.; Sagné, C.; Bellenchi, G.C.; Morel, L.; Darmon, M.; Hůlková, H.; Ruivo, R.; Debacker, C.; El Mestikawy, S.; et al. Expression and Lysosomal Targeting of CLN7, a Major Facilitator Superfamily Transporter Associated with Variant Late-Infantile Neuronal Ceroid Lipofuscinosis. Hum. Mol. Genet. 2010, 19, 4497–4514. [Google Scholar] [CrossRef]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 Are a Common Cause of Variant Late-Infantile Neuronal Ceroid Lipofuscinosis. Brain 2009, 132, 810–819. [Google Scholar] [CrossRef]
- Roosing, S.; van den Born, L.I.; Sangermano, R.; Banfi, S.; Koenekoop, R.K.; Zonneveld-Vrieling, M.N.; Klaver, C.C.W.; van Lith-Verhoeven, J.J.C.; Cremers, F.P.M.; den Hollander, A.I.; et al. Mutations in MFSD8, Encoding a Lysosomal Membrane Protein, Are Associated with Nonsyndromic Autosomal Recessive Macular Dystrophy. Ophthalmology 2015, 122, 170–179. [Google Scholar] [CrossRef]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef]
- Topçu, M.; Tan, H.; Yalnizoğlu, D.; Usubütün, A.; Saatçi, I.; Aynaci, M.; Anlar, B.; Topaloğlu, H.; Turanli, G.; Köse, G.; et al. Evaluation of 36 Patients from Turkey with Neuronal Ceroid Lipofuscinosis: Clinical, Neurophysiological, Neuroradiological and Histopathologic Studies. Turk. J. Pediatr. 2004, 46, 1–10. [Google Scholar]
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 Are a Frequent Cause of Variant-Late Infantile Neuronal Ceroid Lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540. [Google Scholar] [CrossRef]
- Stogmann, E.; El Tawil, S.; Wagenstaller, J.; Gaber, A.; Edris, S.; Abdelhady, A.; Assem-Hilger, E.; Leutmezer, F.; Bonelli, S.; Baumgartner, C.; et al. A Novel Mutation in the MFSD8 Gene in Late Infantile Neuronal Ceroid Lipofuscinosis. Neurogenetics 2009, 10, 73–77. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal Ceroid Lipofuscinosis Caused by MFSD8 Mutations: A Common Theme Emerging. Neurogenetics 2009, 10, 307–311. [Google Scholar] [CrossRef]
- Mandel, H.; Cohen Katsanelson, K.; Khayat, M.; Chervinsky, I.; Vladovski, E.; Iancu, T.C.; Indelman, M.; Horovitz, Y.; Sprecher, E.; Shalev, S.A.; et al. Clinico-Pathological Manifestations of Variant Late Infantile Neuronal Ceroid Lipofuscinosis (VLINCL) Caused by a Novel Mutation in MFSD8 Gene. Eur. J. Med. Genet. 2014, 57, 607–612. [Google Scholar] [CrossRef]
- Patiño, L.C.; Battu, R.; Ortega-Recalde, O.; Nallathambi, J.; Anandula, V.R.; Renukaradhya, U.; Laissue, P. Exome Sequencing Is an Efficient Tool for Variant Late-Infantile Neuronal Ceroid Lipofuscinosis Molecular Diagnosis. PLoS ONE 2014, 9, e109576. [Google Scholar] [CrossRef]
- Kozina, A.A.; Okuneva, E.G.; Baryshnikova, N.V.; Krasnenko, A.Y.; Tsukanov, K.Y.; Klimchuk, O.I.; Kondakova, O.B.; Larionova, A.N.; Batysheva, T.T.; Surkova, E.I.; et al. A Novel MFSD8 Mutation in a Russian Patient with Neuronal Ceroid Lipofuscinosis Type 7: A Case Report. BMC Med. Genet. 2018, 19, 151. [Google Scholar] [CrossRef]
- Bereshneh, A.H.; Garshasbi, M. Novel In-Frame Deletion in MFSD8 Gene Revealed by Trio Whole Exome Sequencing in an Iranian Affected with Neuronal Ceroid Lipofuscinosis Type 7: A Case Report. J. Med. Case Rep. 2018, 12, 281. [Google Scholar] [CrossRef]
- Craiu, D.; Dragostin, O.; Dica, A.; Hoffman-Zacharska, D.; Gos, M.; Bastian, A.E.; Gherghiceanu, M.; Rolfs, A.; Nahavandi, N.; Craiu, M.; et al. Rett-like Onset in Late-Infantile Neuronal Ceroid Lipofuscinosis (CLN7) Caused by Compound Heterozygous Mutation in the MFSD8 Gene and Review of the Literature Data on Clinical Onset Signs. Eur. J. Paediatr. Neurol. 2015, 19, 78–86. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Jilani, A.; Matviychuk, D.; Blaser, S.; Dyack, S.; Mathieu, J.; Prasad, A.N.; Prasad, C.; Kyriakopoulou, L.; Mercimek-Andrews, S. High Diagnostic Yield of Direct Sanger Sequencing in the Diagnosis of Neuronal Ceroid Lipofuscinoses. JIMD Rep. 2019, 50, 20–30. [Google Scholar] [CrossRef]
- Ren, X.-T.; Wang, X.-H.; Ding, C.-H.; Shen, X.; Zhang, H.; Zhang, W.-H.; Li, J.-W.; Ren, C.-H.; Fang, F. Next-Generation Sequencing Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings with Late-Infantile Neuronal Ceroid Lipofuscinosis. Front. Genet. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Kose, M.; Kose, E.; Ünalp, A.; Yılmaz, Ü.; Edizer, S.; Tekin, H.G.; Karaoğlu, P.; Özdemir, T.R.; Er, E.; Onay, H.; et al. Neuronal Ceroid Lipofuscinosis: Genetic and Phenotypic Spectrum of 14 Patients from Turkey. Neurol. Sci. 2021, 42, 1103–1111. [Google Scholar] [CrossRef]
- Rahman, M.M.; Fatema, K. Genetic Diagnosis in Children with Epilepsy and Developmental Disorders by Targeted Gene Panel Analysis in a Developing Country. J. Epilepsy Res. 2021, 11, 22–31. [Google Scholar] [CrossRef]
- Reith, M.; Zeltner, L.; Schäferhoff, K.; Witt, D.; Zuleger, T.; Haack, T.B.; Bornemann, A.; Alber, M.; Ruf, S.; Schoels, L.; et al. A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. Int. J. Mol. Sci. 2022, 23, 2271. [Google Scholar] [CrossRef]
- Poncet, A.F.; Grunewald, O.; Vaclavik, V.; Meunier, I.; Drumare, I.; Pelletier, V.; Bocquet, B.; Todorova, M.G.; Le Moing, A.-G.; Devos, A.; et al. Contribution of Whole-Genome Sequencing and Transcript Analysis to Decipher Retinal Diseases Associated with MFSD8 Variants. Int. J. Mol. Sci. 2022, 23, 4294. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Gong, P.; Jiao, X.; Xu, Z.; Zhang, Y.; Yang, Z. Genetic and Phenotypic Spectrum of Chinese Patients with Epilepsy and Photosensitivity. Front. Neurol. 2022, 13, 907228. [Google Scholar] [CrossRef]
- Sürücü Kara, İ.; Köse, E.; Doğulu, N.; Yüksel, M.F.; Ceylaner, S.; Kendirli, T.; Eminoğlu, F.T. Severe Rhabdomyolysis in Neuronal Ceroid Lipofuscinosis Type 7. Clin. Neurol. Neurosurg. 2022, 220, 107375. [Google Scholar] [CrossRef]
- Qiao, Y.; Gu, Y.; Cheng, Y.; Su, Y.; Lv, N.; Shang, Q.; Xing, Q. Case Report: Novel MFSD8 Variants in a Chinese Family with Neuronal Ceroid Lipofuscinoses 7. Front. Genet. 2022, 13, 807515. [Google Scholar] [CrossRef]
- Vreeswijk, M.P.G.; van der Klift, H.M. Analysis and Interpretation of RNA Splicing Alterations in Genes Involved in Genetic Disorders. In Exon Skipping: Methods and Protocols; Aartsma-Rus, A., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 49–63. ISBN 978-1-61779-767-5. [Google Scholar]
- Chomczynski, P.; Sacchi, N. The Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate–Phenol–Chloroform Extraction: Twenty-Something Years On. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Steenhuis, P.; Froemming, J.; Reinheckel, T.; Storch, S. Proteolytic Cleavage of the Disease-Related Lysosomal Membrane Glycoprotein CLN7. Biochim. Biophys. Acta 2012, 1822, 1617–1628. [Google Scholar] [CrossRef]
- Priluck, A.Z.; Breazzano, M.P. Novel MFSD8 Mutation Causing Non-Syndromic Asymmetric Adult-Onset Macular Dystrophy. Ophthalmic Genet. 2022, 43, 1–5. [Google Scholar] [CrossRef]
- Zare-Abdollahi, D.; Bushehri, A.; Alavi, A.; Dehghani, A.; Mousavi-Mirkala, M.; Effati, J.; Miratashi, S.A.M.; Dehani, M.; Jamali, P.; Khorshid, H.R.K. MFSD8 Gene Mutations; Evidence for Phenotypic Heterogeneity. Ophthalmic Genet. 2019, 40, 141–145. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-Generation Sequencing Identifies Unexpected Genotype-Phenotype Correlations in Patients with Retinitis Pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef]
- Geier, E.G.; Bourdenx, M.; Storm, N.J.; Cochran, J.N.; Sirkis, D.W.; Hwang, J.-H.; Bonham, L.W.; Ramos, E.M.; Diaz, A.; Van Berlo, V.; et al. Rare Variants in the Neuronal Ceroid Lipofuscinosis Gene MFSD8 Are Candidate Risk Factors for Frontotemporal Dementia. Acta Neuropathol. 2019, 137, 71–88. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Nucleotide Change [NM_001371596.2] | Protein Change [NP_001358525.1] | [Reference]: n. of Patients- zygosity | LINCL | NS- RD * | Other |
|---|---|---|---|---|---|
| c.2T > C | p.? | [13]: 1 ComHet | 1 | ||
| c.103C > T | p.(Arg35 *) | [9]: 1 Homo, [11]: 1 ComHet, [6]: 1 Homo + 1 ComHet, [13]: 1 Homo | 4 | 1 | |
| c.[104G > A;155G > C] | p.[(Arg35Gln);(Gly52Ala)] | [27]: 1 ComHet | 1 | ||
| c.136_137del | p.(Met46Valfs * 22) | [30]: 1 ComHet | 1 | ||
| c.154G > A | p.(Gly52Arg) | [13]: 1 ComHet [34]: 1 ComHet | 1 | 1 | |
| c.233G > A | p.(Trp78 *) | [11]: 2 ComHet | 2 (2) | ||
| c.259C > T | p.(Gln87 *) | [6]: 1 Homo | 1 | ||
| c.301G > C | p.(Ala146Pro) | [24]: 1 Homo | 1 | ||
| c.325_339del | p.(Val109_Ile113del) | [19]: 1 Homo | 1 | ||
| c.362A > G | p.(Tyr121Cys) | [14]: 5 Homo | 5 (5) | ||
| c.416G > A | p.(Arg139His) | [9]: 1 Homo, [22]: 1 Homo, [6]: 1 Homo | 3 | ||
| c.468_469delinsCC | p.(Ala157Pro) | [9]: 1 Homo | jNCL | ||
| c.472G > A | p.(Gly158Ser) | [16]: 5 Homo | 5 (5) | ||
| c.479C > A | p.(Thr160Asn) | [6]: 1 Homo | 1 | ||
| c.479C > T | p.(Thr160Ile) | [6]: 1 ComHet | 1 | ||
| c.525T > A | p.(Cys175*) | [18]: 1 Homo | 1 | ||
| c.554-1G > C | p.? | [6]: 1 ComHet | 1 | ||
| c.554-5A > G | p.? | [23]: 1 ComHet | 1 | ||
| SVA insertion | p.[Val185Aspfs * 3,?] | [21]: 1 ComHet | 1 | ||
| c.557T > G | p.(Phe186Cys) | [28]: 1 ComHet | 1 | ||
| c.627_643del | p.(Met209Ilefs * 3) | [9]: 1 Homo, [13]: 1 Homo | 1 | ||
| c.679A > G | p.(Arg233Gly) | [12]: 1 Homo | 1 | ||
| c.721G > T | p.(Gly241 *) | [24]: 1 Homo | 1 | ||
| c.750A > G | p.[Arg233Serfs * 5,=] | [26]: 2 Homo, [27]: 2 ComHet | 1 | 1 | 2 jNCL |
| c.754 + 1G > A | p.? | [6]: 1 Homo, 1 ComHet | 2 | ||
| c.754 + 2T > A | p.[Arg233Serfs * 5,=] | [6]: 6 Homo 2 + ComHet, [7]: 1 Homo, [12]: 1 Homo, [20]: 1ComHet, [22]: 1 Homo | 12 (2) | ||
| c.755- 2726_998 + 1981delinsGTA | p.(Ala252Glyfs * 82) | [27]: 1 ComHet | 1 | ||
| c.850G > C | p.(Ala284Pro) | [25]: 1 Homo | 1 | ||
| c.863 + 1G > C | p.? | [9]: 1 Homo, [13]: 1 Homo | 2 | ||
| c.863 + 3_863 + 4insT | p.? | [13]: 3 ComHet | 3 | ||
| c.863 + 4A > G | p.? | [22]: 1 Homo | 1 | ||
| c.881C > A | p.(Thr294Lys) | [9]: 18 Homo, [6]: 3 Homo + 1 ComHet, [13]: 1 ComHet, [20]: 1 ComHet, [22]: 1 Homo, [27]: 2 ComHet | 25 (2,2,2) | 2 | |
| c.894T > G | p.(Tyr298 *) | [7]: 1 Homo | 1 | ||
| c.929G > A | p.(Gly310Asp) | [7]: 1 Homo, [13]: 1 ComHet, [6]: 1 Homo, [27]: 1 ComHet | 3 | 1 | |
| c.998 + 1669A > G | p.[=,Lys333Asnfs * 18] | [27]: 1 ComHet | 1 | ||
| c.1006C > G | p.(Glu336Gln) | [10]: 6 ComHet, [11]: 6 ComHet, [34]: 1 ComHet, [27]: 2 ComHet | 13 (5,3,2) | ||
| c.1006G > A | p.(Glu336Lys) | [27]: 1 ComHet | 1 | ||
| c.1009C > T | p.(Arg337Cys) | [27]: 1 ComHet | 1 | ||
| c.1093C > T | p.(Gln365 *) | [24]: 2 Homo | 2 | ||
| c.1102G > C | p.[Asp368His, Ile334Phefs * 4] | [7]: 1 Homo, [21]: 1ComHet, [10]: 1 ComHet | 2 | 1 | |
| c.1103-2del | p.? | [9]: 1 ComHet | 1 | ||
| c.1141G > T | p.(Glu381 *) | [13]: 1 ComHet, [10]: 5 ComHet, [27]: 1 ComHet | 1 | 6 (5) | |
| c.1219T > C | p.(Trp407Arg) | [17]: 3 ComHet | 3 | ||
| c.1235C > T | p.(Pro412Leu) | [15]: 3 Homo, [6]: 1 ComHet, [35]: 3 ComHet | 4 (3) | 3 | |
| c.1241_1242ins GAAT | p.(Ile414Metfs * 109) | [22]: 1 Homo | 1 | ||
| c.1265C > A | p.(Ser422 *) | [27]: 1 ComHet | 1 | ||
| c.1286G > A | p.(Gly429Asp) | [7]: 1 Homo | 1 | ||
| c.1340C > T | p.(Pro447Leu) | [13]: 1 Homo | 1 | ||
| c.1351-1G > A | p.? | [28]: 1 ComHet | 1 | ||
| c.1361T > C | p.(Met454Thr) | [17]: 3 ComHet, [11]: 6 Homo, [35]: 3 ComHet, 2 Homo | 3 | 11 (3,2) | |
| c.1373C > A | p.(Thr458Lys) | [6]: 1 ComHet | 1 | ||
| c.1393C > T | p.(Arg465Trp) | [9]: 1Homo | 1 | ||
| c.1394G > A | p.(Arg465Gln) | [6]: 1 ComHet, [22]: 1 Homo, [11]: 3 ComHet | 2 | 3 (3) | |
| c.1408A > G | p.(Met470Val) | [6]: 1 ComHet | 1 | ||
| c.1420C > T | p.(Gln474 *) | [6]: 1 Homo | 1 | ||
| c.1444C > T | p.(Arg482 *) | [13]: 1 ComHet, [6]: 1 ComHet, [23]: 1 ComHet, | 3 | ||
| c.1445G > C | p.(Arg482Pro) | [36]: 3 Homo | 3 (3) | ||
| Deletion exon1–4 | p.? | [29]: 1 Homo | 1 | ||
| Whole gene deletion (180 kb) | [30]: 1 ComHet | 1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquetti, D.; Marangi, G.; Orteschi, D.; Carapelle, M.; L’Erario, F.F.; Venditti, R.; Maietta, S.; Battaglia, D.I.; Contaldo, I.; Veredice, C.; et al. Linear Diagnostic Procedure Elicited by Clinical Genetics and Validated by mRNA Analysis in Neuronal Ceroid Lipofuscinosis 7 Associated with a Novel Non-Canonical Splice Site Variant in MFSD8. Genes 2023, 14, 245. https://doi.org/10.3390/genes14020245
Pasquetti D, Marangi G, Orteschi D, Carapelle M, L’Erario FF, Venditti R, Maietta S, Battaglia DI, Contaldo I, Veredice C, et al. Linear Diagnostic Procedure Elicited by Clinical Genetics and Validated by mRNA Analysis in Neuronal Ceroid Lipofuscinosis 7 Associated with a Novel Non-Canonical Splice Site Variant in MFSD8. Genes. 2023; 14(2):245. https://doi.org/10.3390/genes14020245
Chicago/Turabian StylePasquetti, Domizia, Giuseppe Marangi, Daniela Orteschi, Marina Carapelle, Federica Francesca L’Erario, Romina Venditti, Sabrina Maietta, Domenica Immacolata Battaglia, Ilaria Contaldo, Chiara Veredice, and et al. 2023. "Linear Diagnostic Procedure Elicited by Clinical Genetics and Validated by mRNA Analysis in Neuronal Ceroid Lipofuscinosis 7 Associated with a Novel Non-Canonical Splice Site Variant in MFSD8" Genes 14, no. 2: 245. https://doi.org/10.3390/genes14020245
APA StylePasquetti, D., Marangi, G., Orteschi, D., Carapelle, M., L’Erario, F. F., Venditti, R., Maietta, S., Battaglia, D. I., Contaldo, I., Veredice, C., & Zollino, M. (2023). Linear Diagnostic Procedure Elicited by Clinical Genetics and Validated by mRNA Analysis in Neuronal Ceroid Lipofuscinosis 7 Associated with a Novel Non-Canonical Splice Site Variant in MFSD8. Genes, 14(2), 245. https://doi.org/10.3390/genes14020245