
Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men

 , and
, and 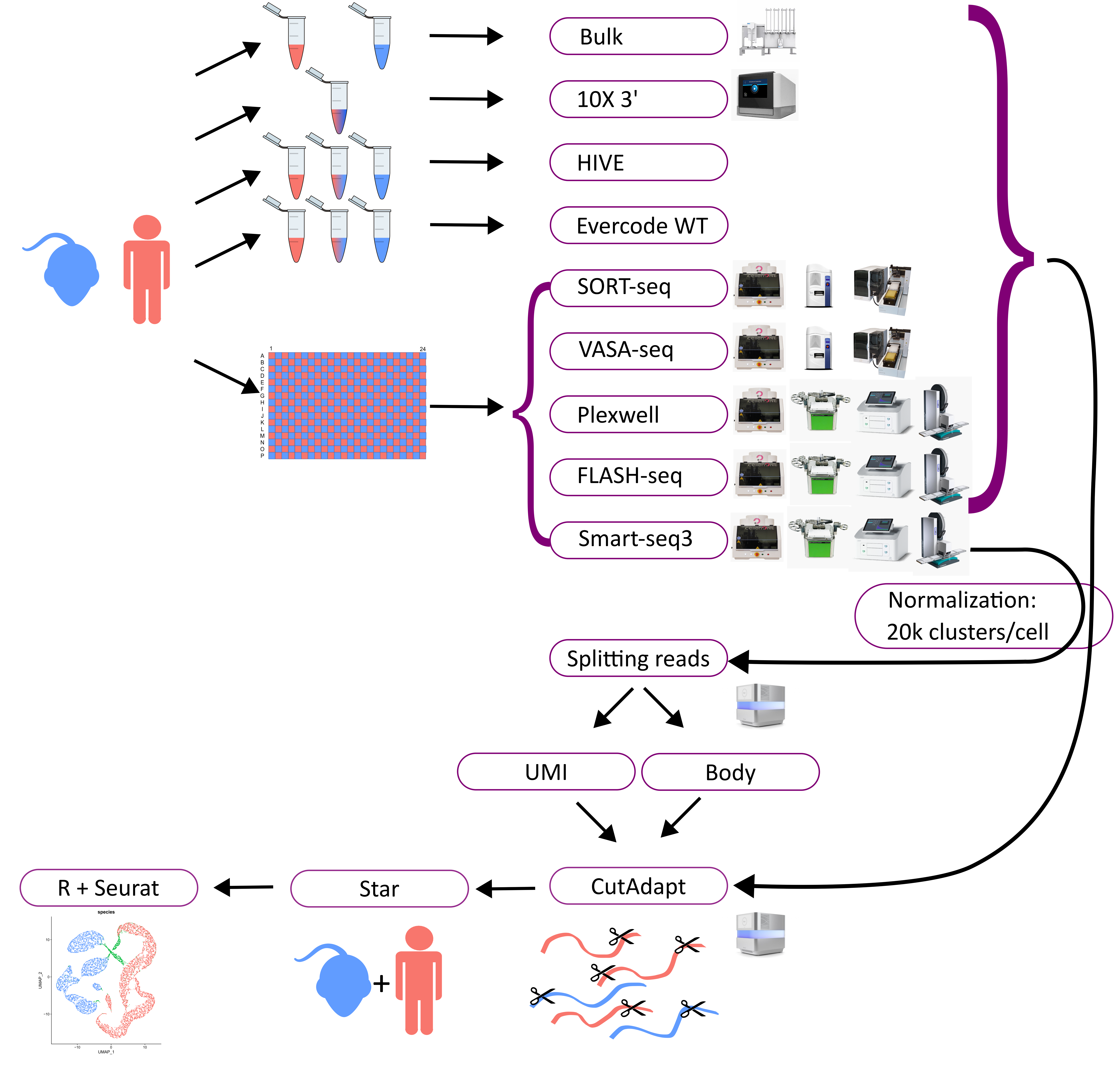{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Growth and Sorting
2.2. Single Cell Transcriptome Assays
2.3. Bulk RNAseq
2.4. Sequencing
2.5. Data Analysis
3. Results
3.1. Single Cell RNAseq Requirements
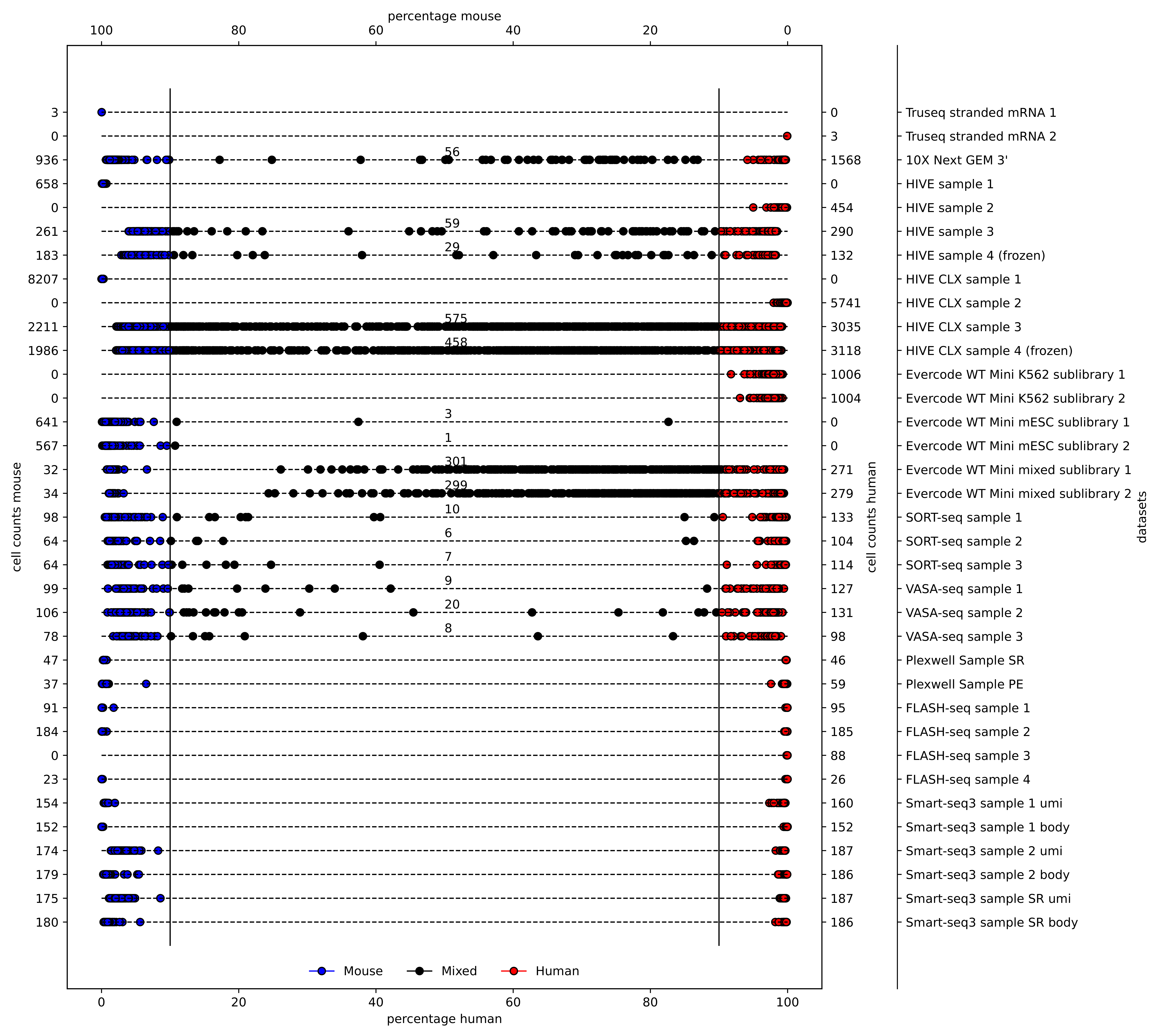
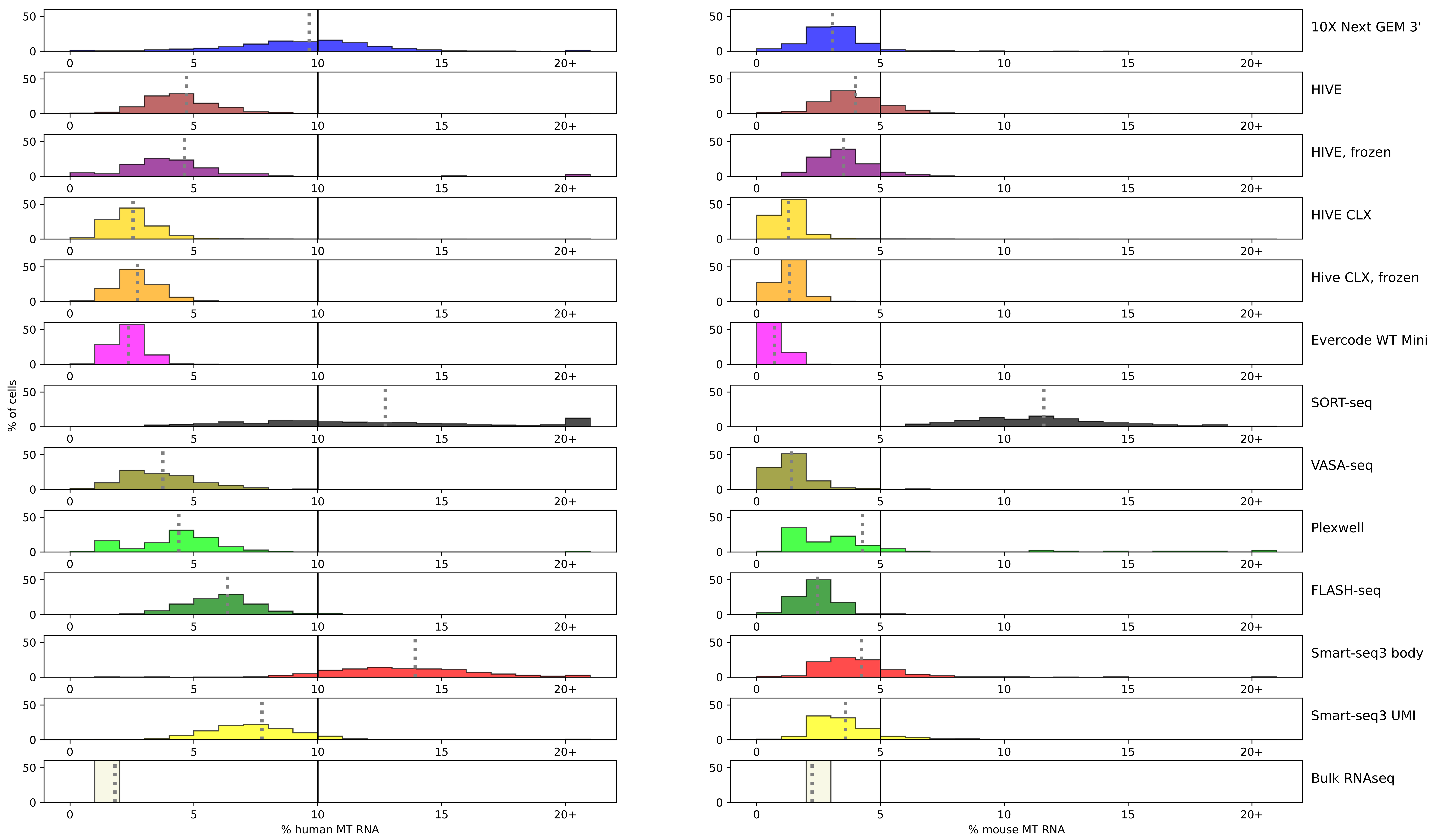
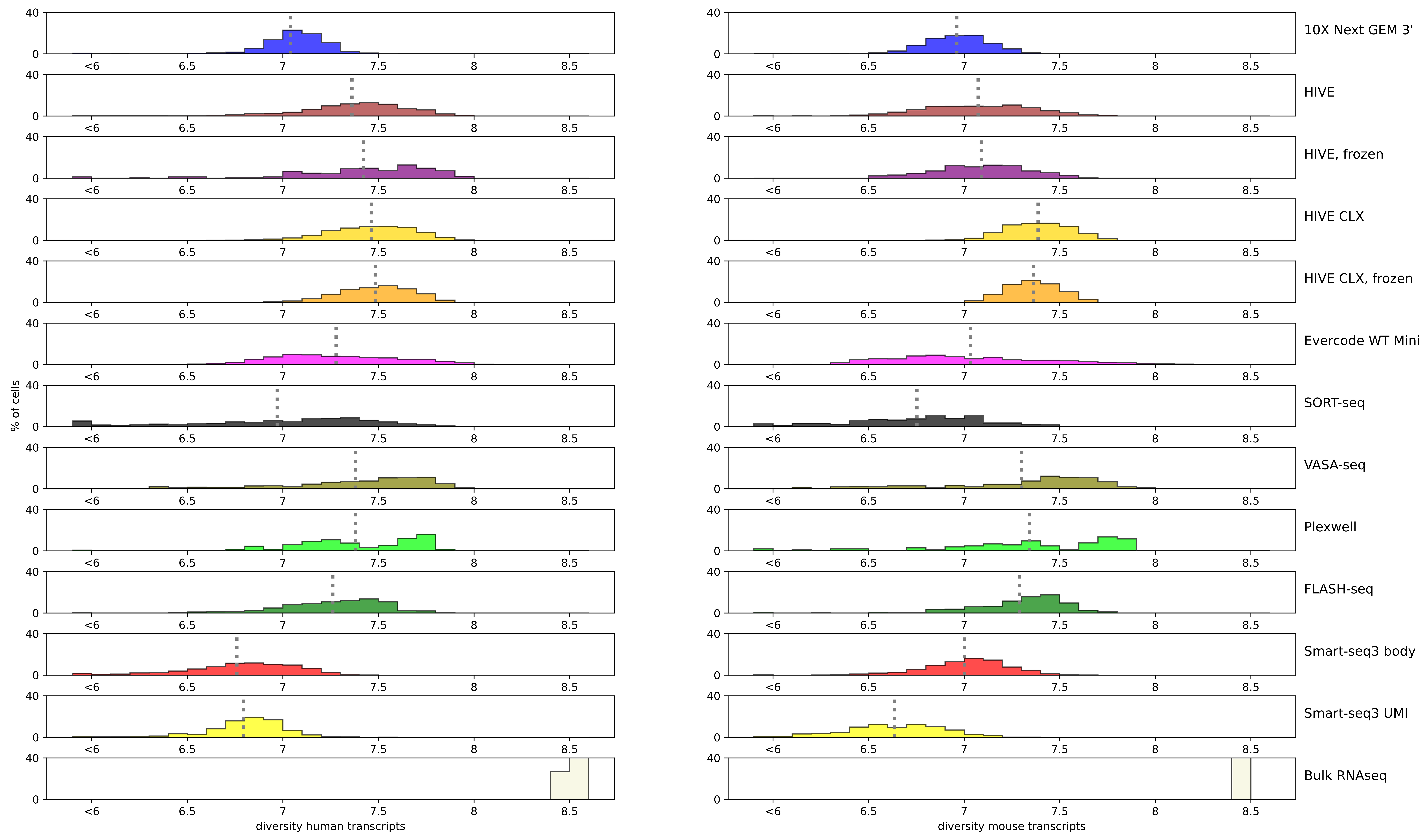
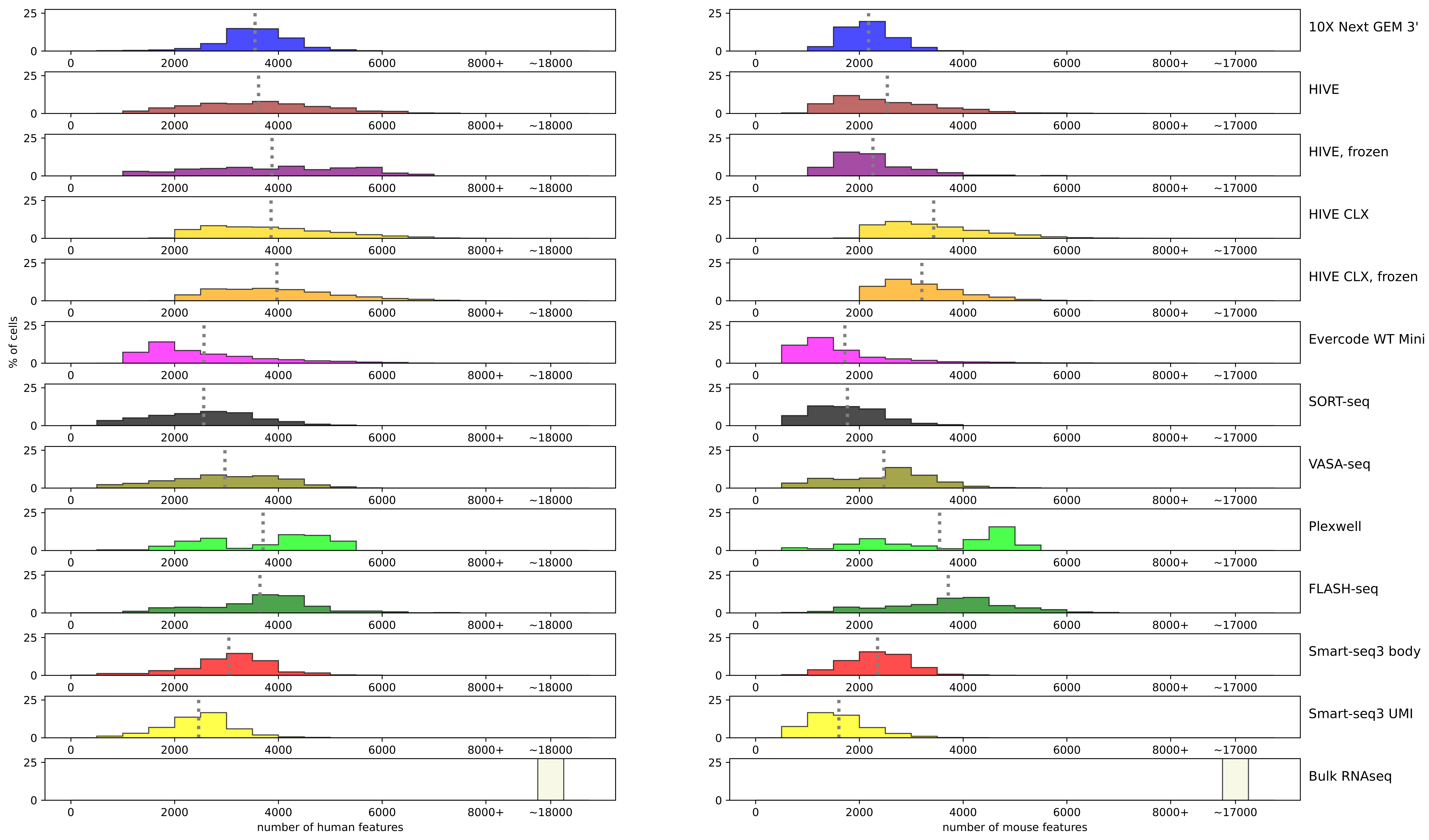
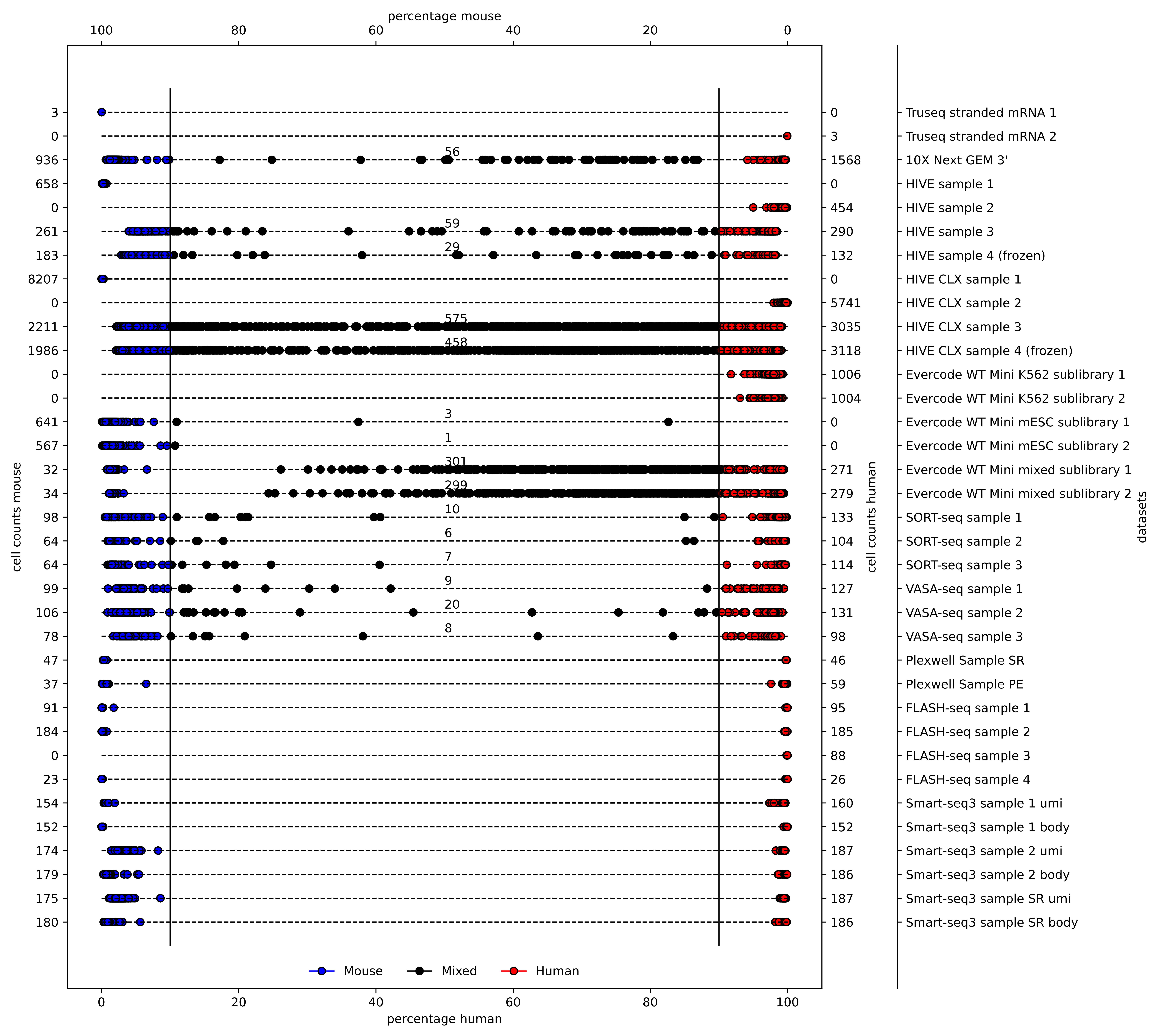
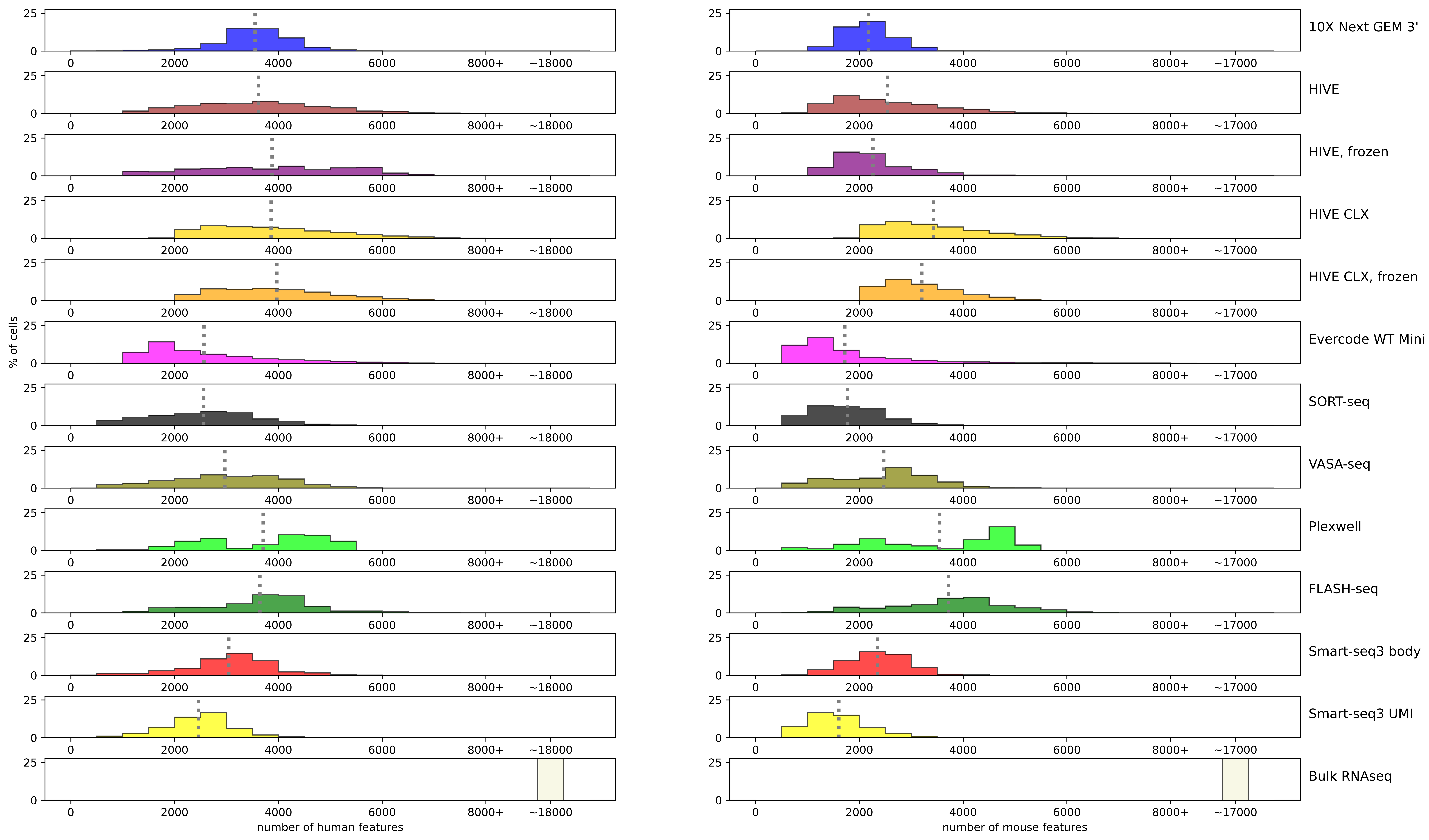
3.2. Quality Control
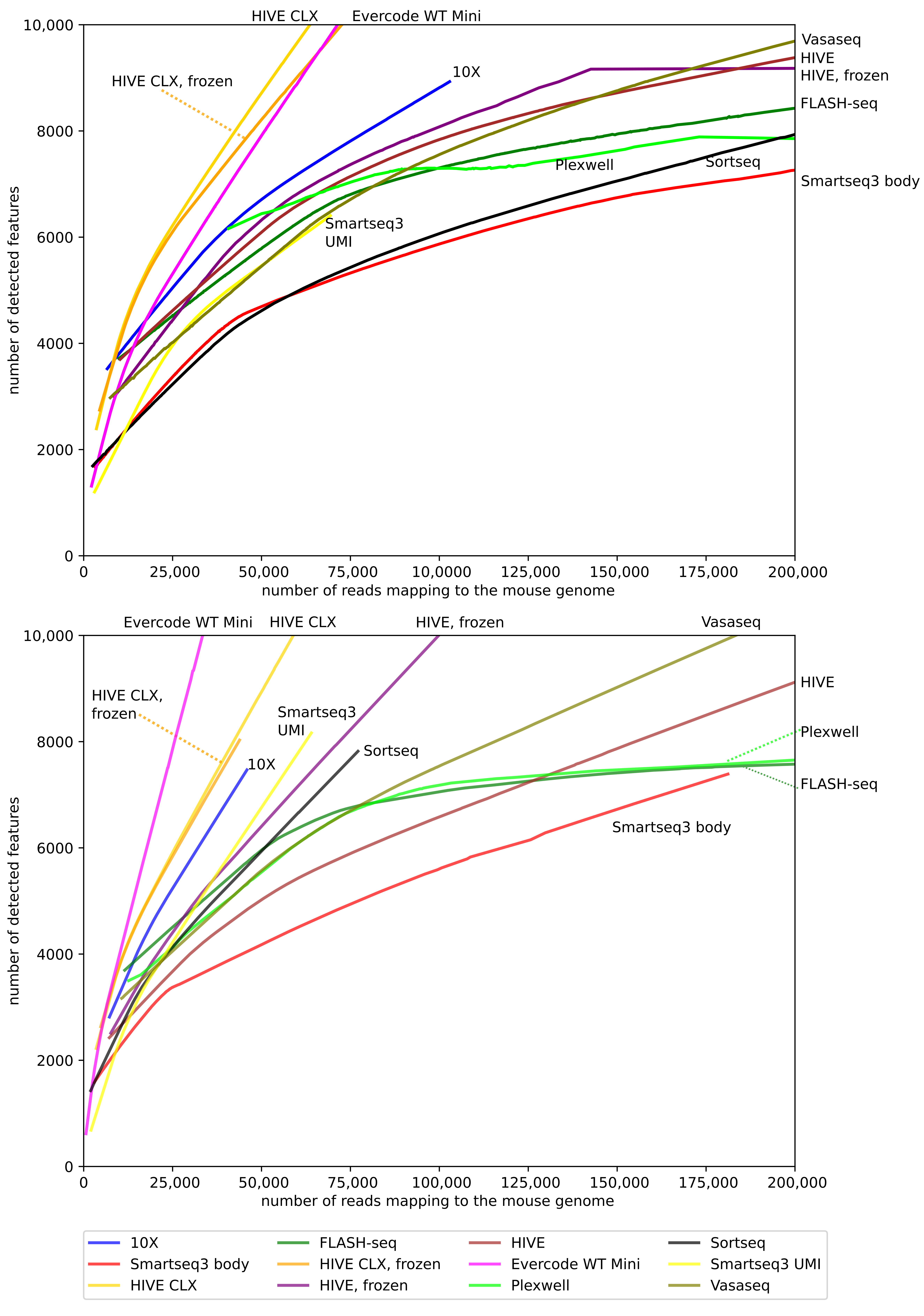
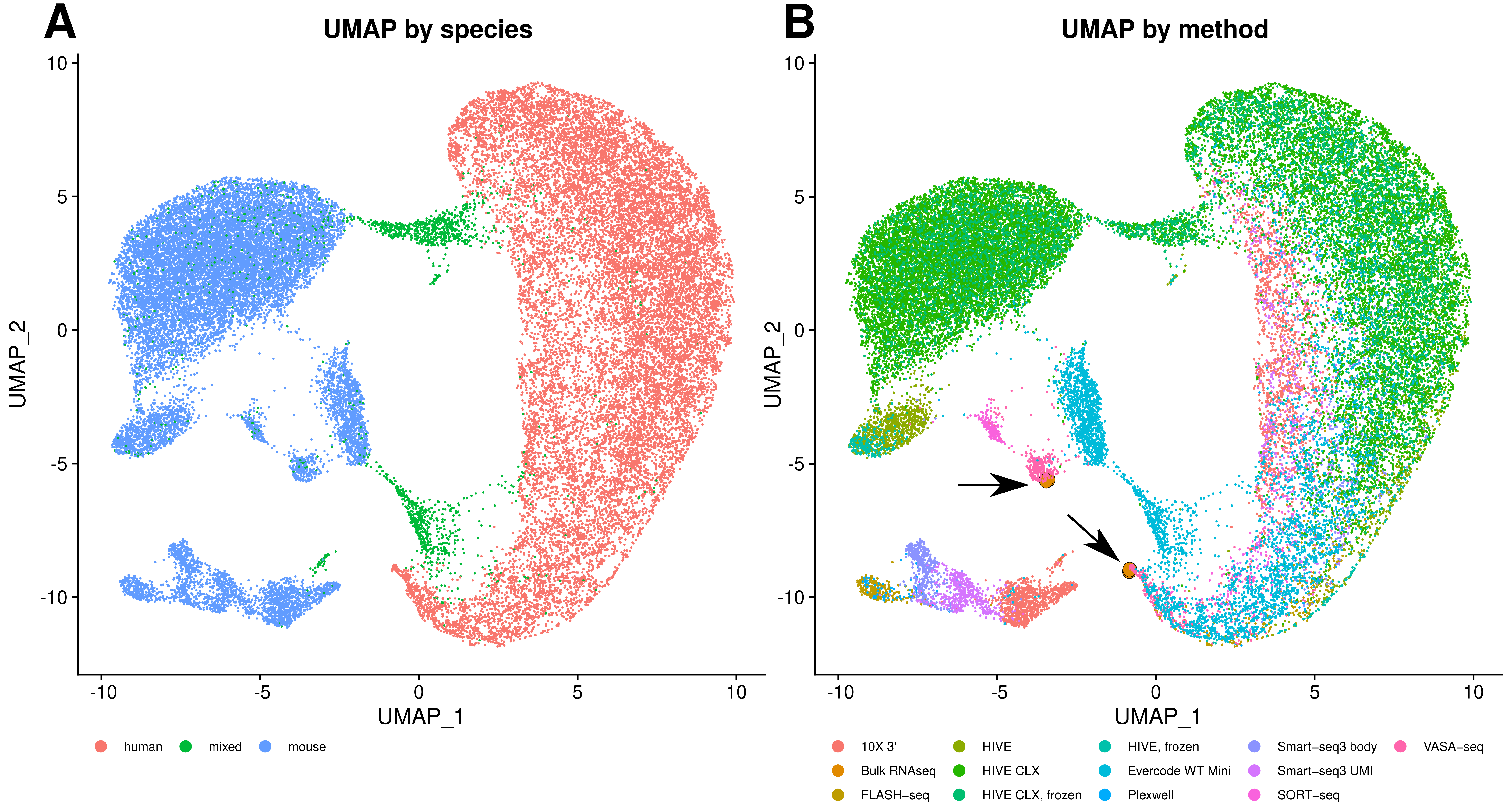
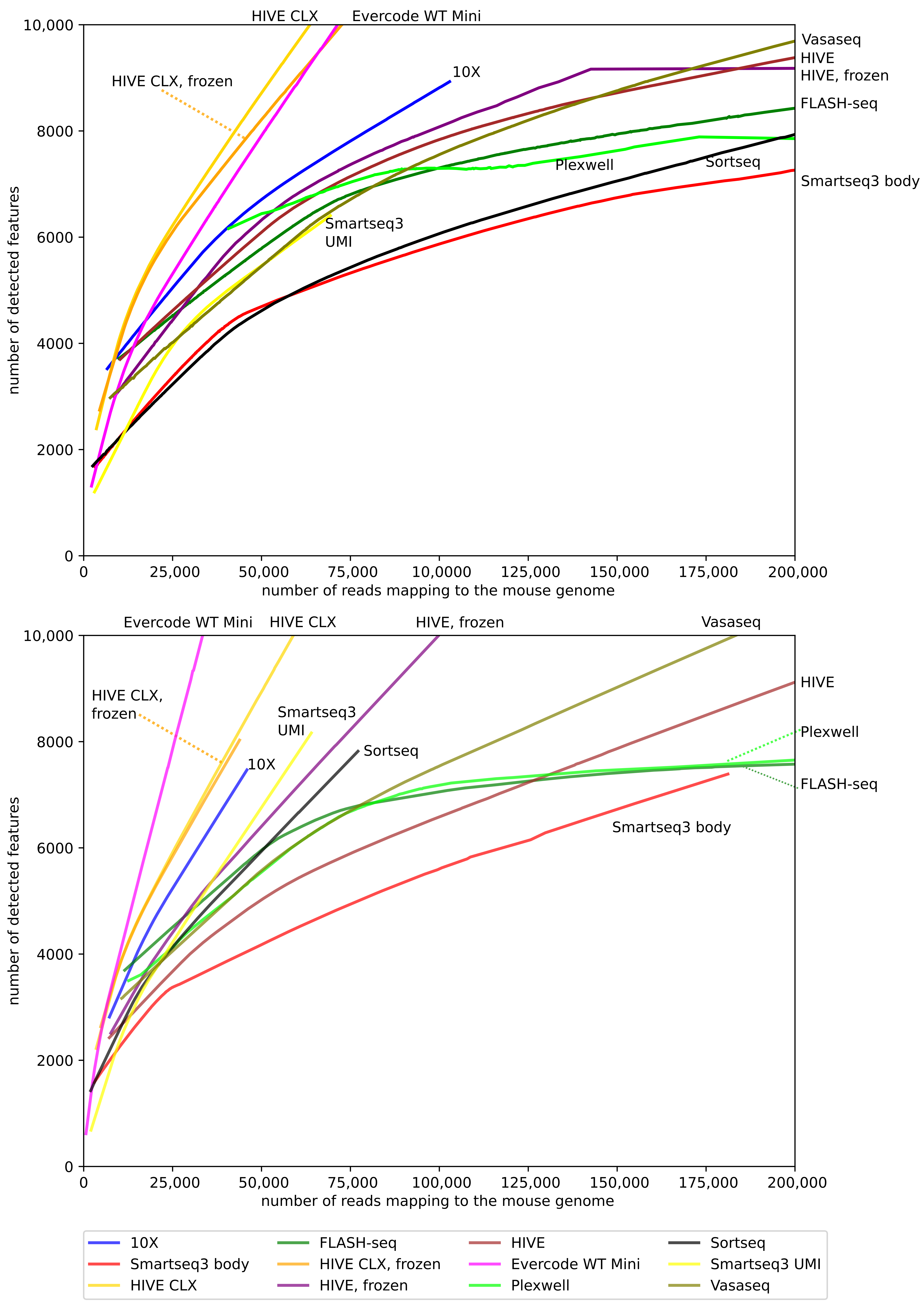
3.3. Performance of the Single Cell Methods
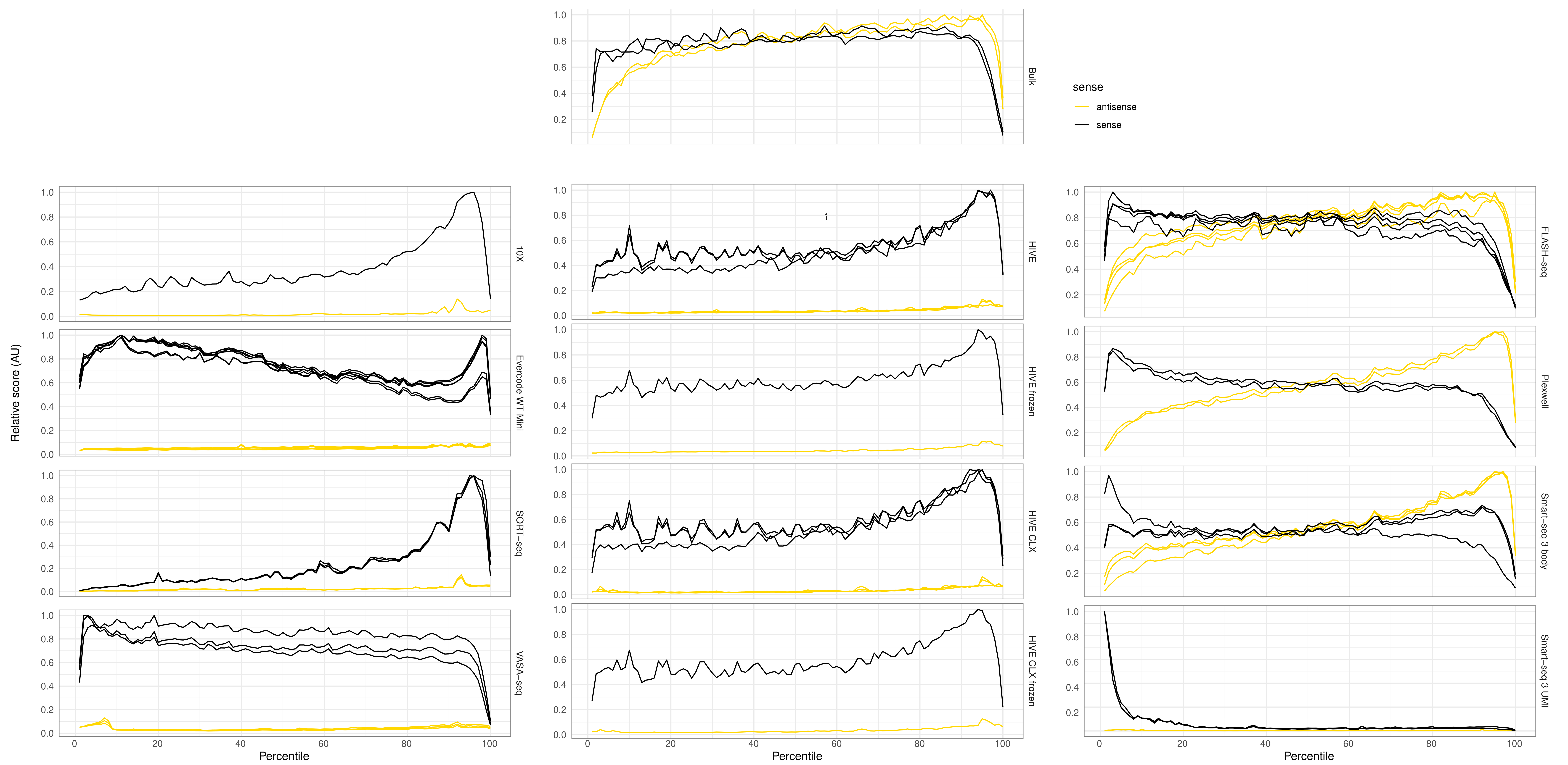
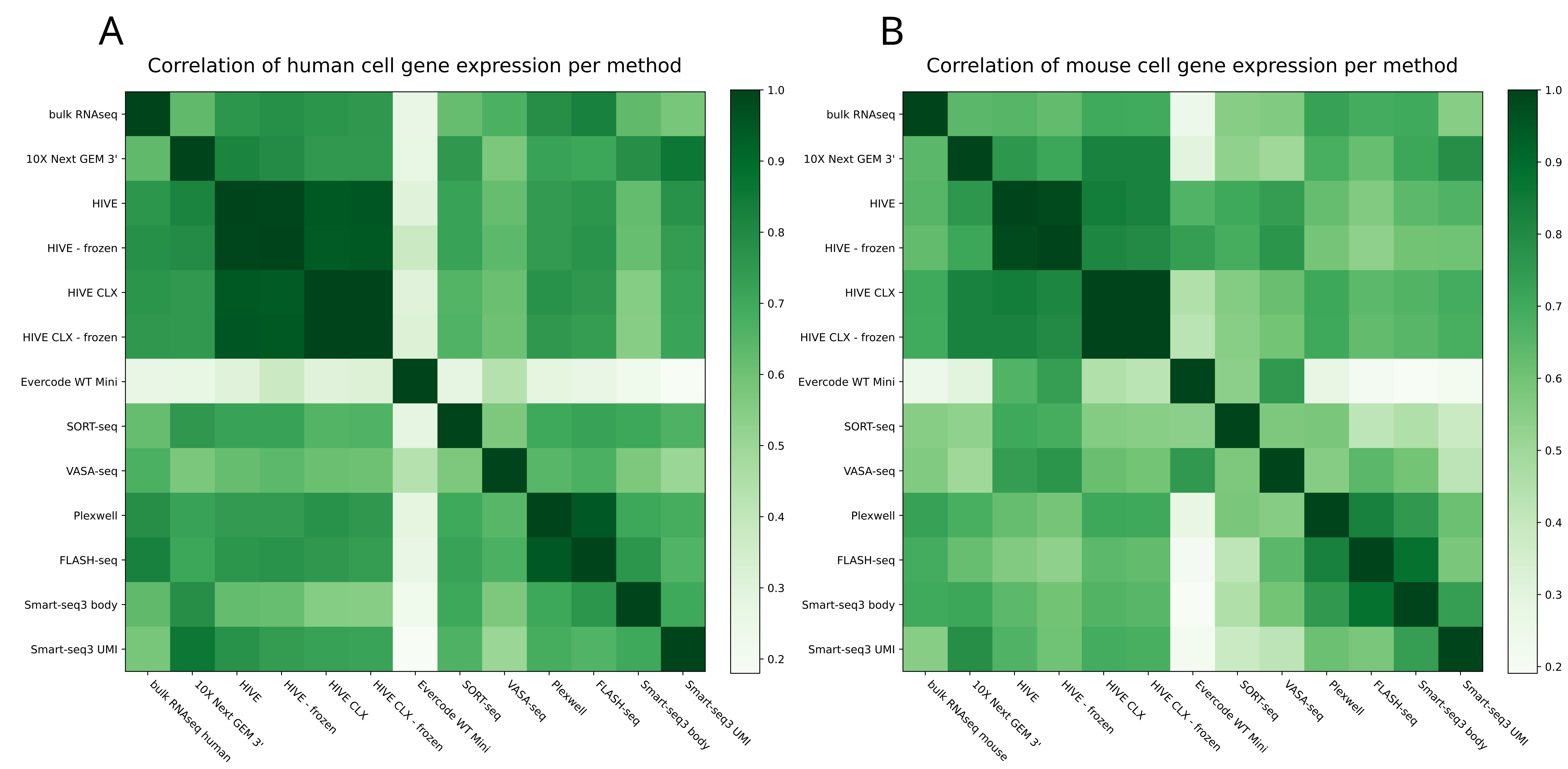
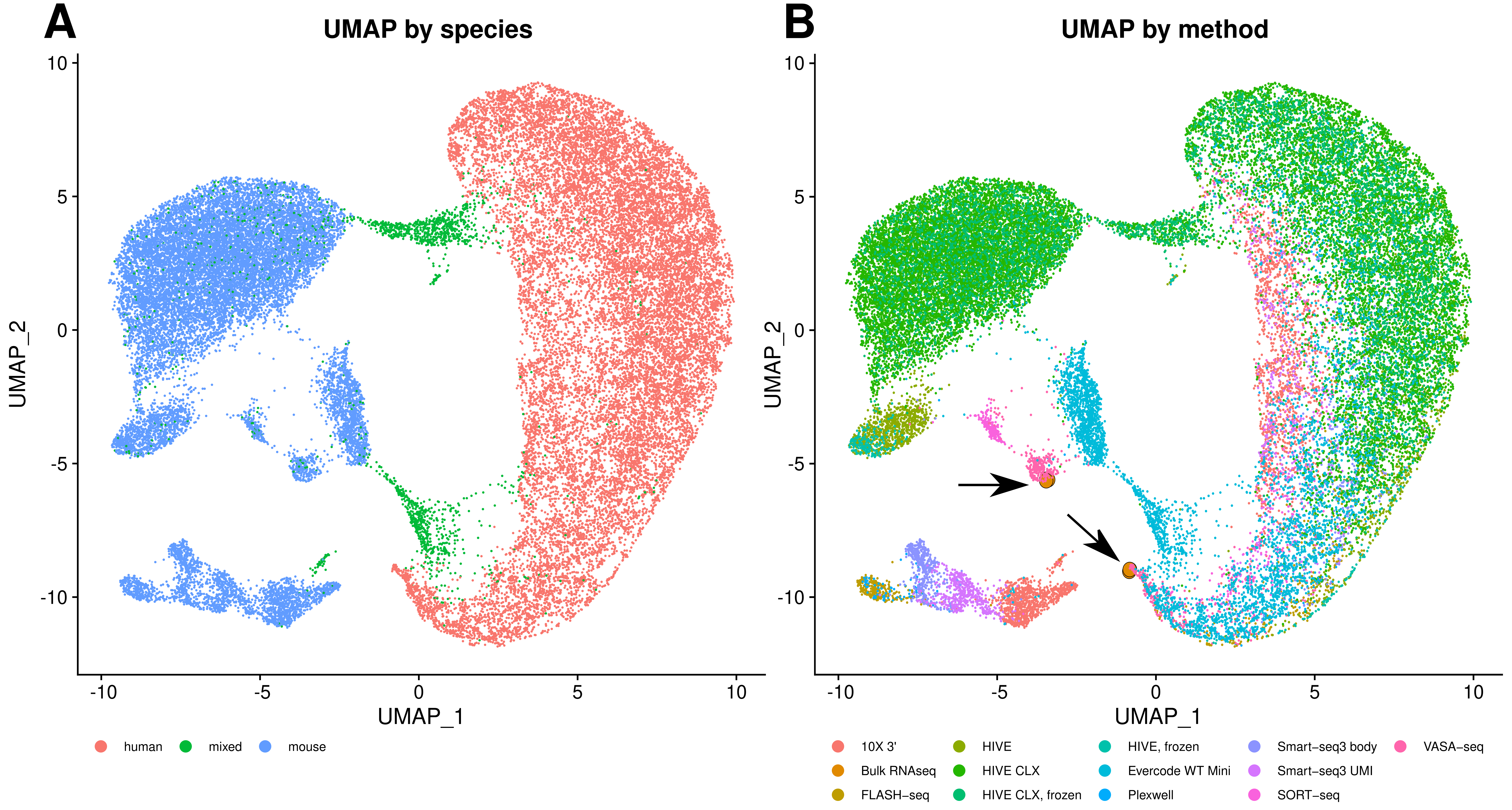
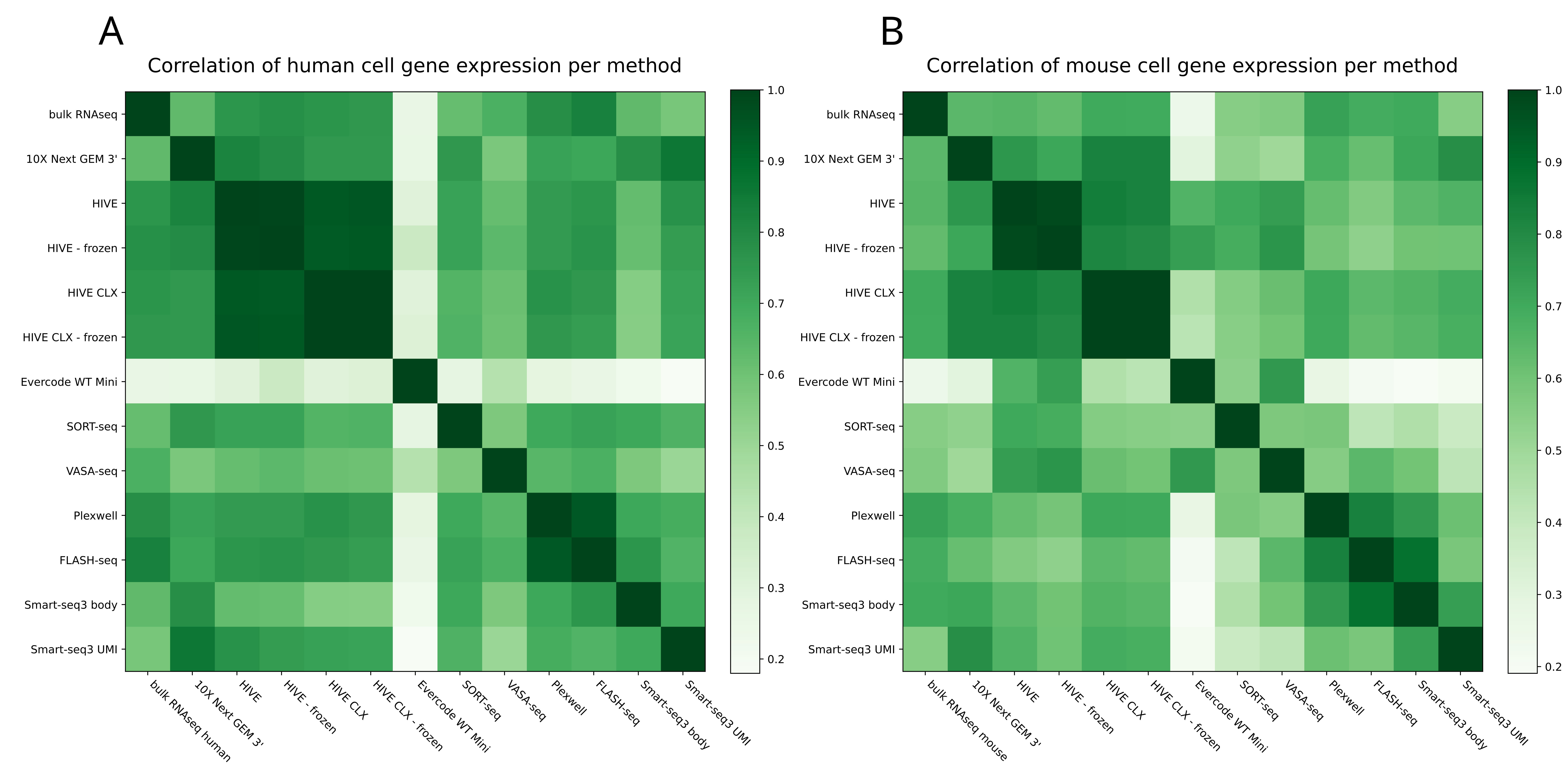
3.4. Comparability of Profiles
4. Discussion
4.1. Time Requirements and Automation
4.2. Filtering Cells
4.3. Multiplets
4.4. Batch Effect
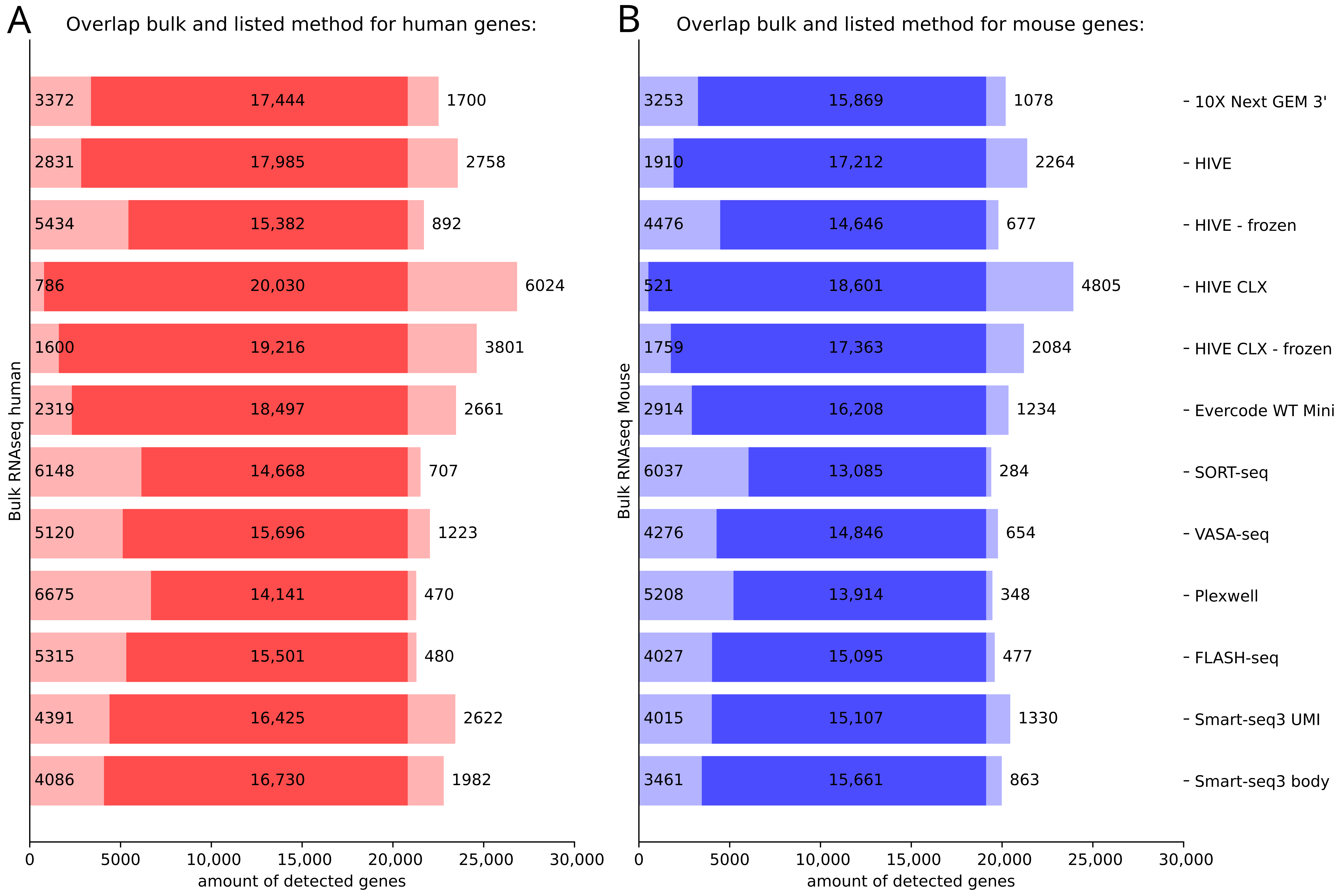
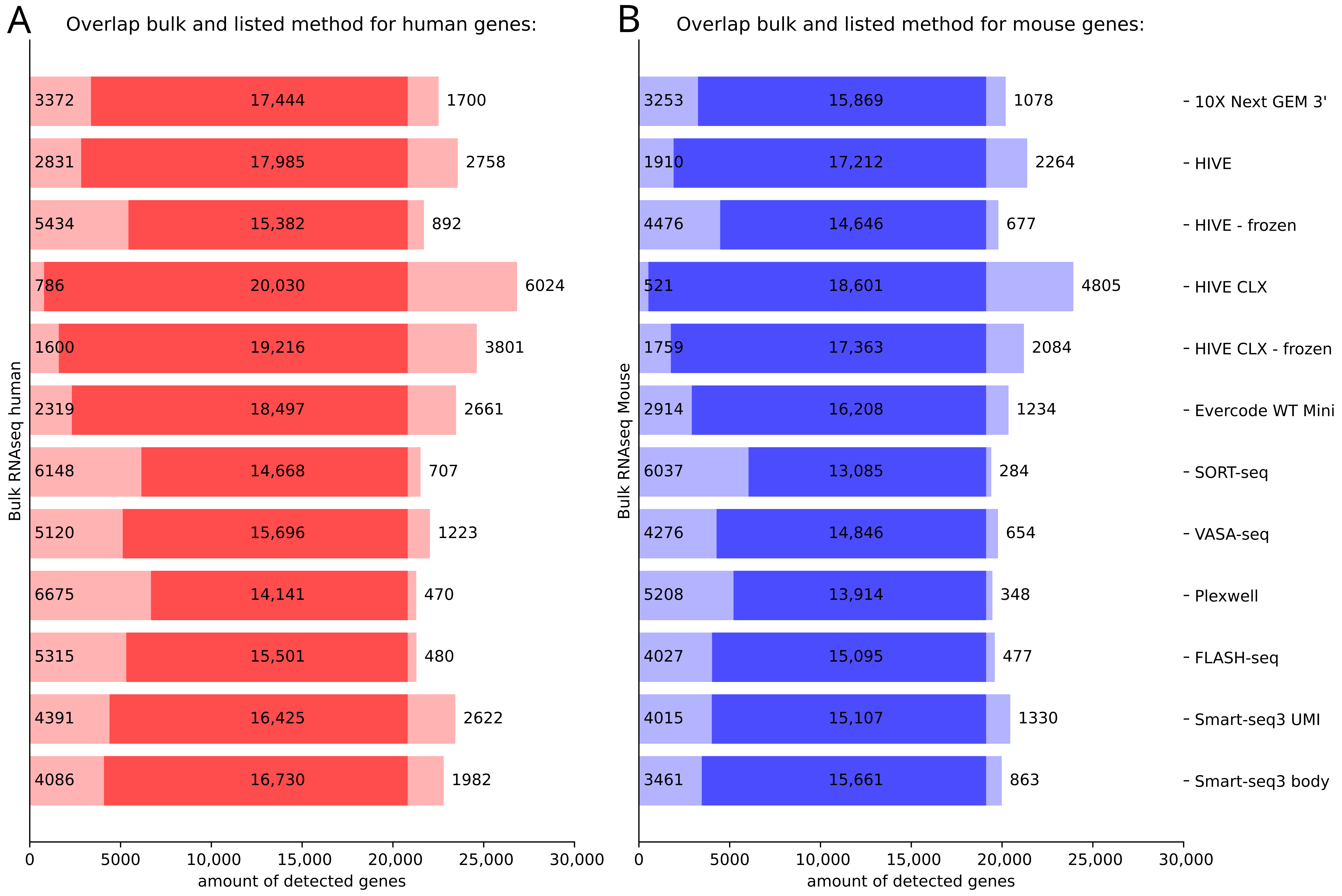
4.5. Overlap and Difference between the Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Method of the Year 2013. Nat. Methods 2014, 11, 1. [CrossRef] [PubMed]
- Chi, K.R. Singled out for sequencing. Nat. Methods 2014, 11, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods 2014, 11, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.-Y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E.; et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Neagu, A.; van Genderen, E.; Escudero, I.; Verwegen, L.; Kurek, D.; Lehmann, J.; Stel, J.; Dirks, R.A.M.; van Mierlo, G.; Maas, A.; et al. In vitro capture and characterization of embryonic rosette-stage pluripotency between naive and primed states. Nat. Cell Biol. 2020, 22, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramskold, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramskold, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Smart-seq3 Protocol; Version 3; Protocols.io: Berkeley, CA, USA, 2022. [Google Scholar] [CrossRef]
- Hahaut, V.; Pavlinic, D.; Carbone, W.; Schuierer, S.; Balmer, P.; Quinodoz, M.; Renner, M.; Roma, G.; Cowan, C.S.; Picelli, S. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat. Biotechnol. 2022, 40, 1447–1451. [Google Scholar] [CrossRef]
- Picelli, S.; Hahaut, V. FLASH-seq Protocol, version 3. Protocols.io: Berkeley, CA, USA. 2022. Available online: https://www.protocols.io/view/flash-seq-protocol-b6myrc7w.html (accessed on 20 June 2023).
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Salmen, F.; De Jonghe, J.; Kaminski, T.S.; Alemany, A.; Parada, G.E.; Verity-Legg, J.; Yanagida, A.; Kohler, T.N.; Battich, N.; van den Brekel, F.; et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 2022, 40, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.J.; Dharmadhikari, G.; Grun, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394. [Google Scholar] [CrossRef]
- van den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Molder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable data analysis with Snakemake. F1000Res 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Church, D.M.; Goodstadt, L.; Hillier, L.W.; Zody, M.C.; Goldstein, S.; She, X.; Bult, C.J.; Agarwala, R.; Cherry, J.L.; DiCuccio, M.; et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009, 7, e1000112. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Seabold, S.; Perktold, J. statsmodels: Econometric and statistical modeling with python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Harrington, B.; Inkscape Contributors. The Inkscape Project. Available online: https://inkscape.org/about/ (accessed on 25 September 2023).
- Osorio, D.; Cai, J.J. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics 2021, 37, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lonnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Ziegenhain, C.; Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. 2022, 40, 1452–1457. [Google Scholar] [CrossRef]
- Xi, N.M.; Li, J.J. Benchmarking Computational Doublet-Detection Methods for Single-Cell RNA Sequencing Data. Cell Syst. 2021, 12, 176–194. [Google Scholar] [CrossRef]
- Why is the Multiplet Rate Different for the Next GEM Single Cell 3’ LT v3.1 Assay Compared to Other Single Cell Applications? Available online: https://kb.10xgenomics.com/hc/en-us/articles/360059124751-Why-is-the-multiplet-rate-different-for-the-Next-GEM-Single-Cell-3-LT-v3-1-assay-compared-to-other-single-cell-applications (accessed on 20 June 2023).
- Lareau, C.A.; Ma, S.; Duarte, F.M.; Buenrostro, J.D. Inference and effects of barcode multiplets in droplet-based single-cell assays. Nat. Commun. 2020, 11, 866. [Google Scholar] [CrossRef]
- What Is the Doublet Rate? Available online: https://honeycomb.bio/faqs/ (accessed on 27 June 2023).
- Parse Biosciences. Performance of Evercode™ WT in a Multi-Species Cell Line Experiment. Available online: https://www.parsebiosciences.com/datasets/cell-line/performance-of-evercode-wt-in-a-multi-species-cell-line-experiment (accessed on 31 October 2023).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hornung, B.V.H.; Azmani, Z.; den Dekker, A.T.; Oole, E.; Ozgur, Z.; Brouwer, R.W.W.; van den Hout, M.C.G.N.; van IJcken, W.F.J. Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men. Genes 2023, 14, 2226. https://doi.org/10.3390/genes14122226
Hornung BVH, Azmani Z, den Dekker AT, Oole E, Ozgur Z, Brouwer RWW, van den Hout MCGN, van IJcken WFJ. Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men. Genes. 2023; 14(12):2226. https://doi.org/10.3390/genes14122226
Chicago/Turabian StyleHornung, Bastian V. H., Zakia Azmani, Alexander T. den Dekker, Edwin Oole, Zeliha Ozgur, Rutger W. W. Brouwer, Mirjam C. G. N. van den Hout, and Wilfred F. J. van IJcken. 2023. "Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men" Genes 14, no. 12: 2226. https://doi.org/10.3390/genes14122226
APA StyleHornung, B. V. H., Azmani, Z., den Dekker, A. T., Oole, E., Ozgur, Z., Brouwer, R. W. W., van den Hout, M. C. G. N., & van IJcken, W. F. J. (2023). Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men. Genes, 14(12), 2226. https://doi.org/10.3390/genes14122226