
The Genome of the Yellow Mealworm, Tenebrio molitor: It’s Bigger Than You Think

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Insects
2.2. Genome Sequencing and Assembly
2.3. Gene Prediction, Annotation, and Analysis
2.4. Transcriptome Sequencing and Analysis
2.5. Satellite DNA (SatDNA) Detection and Analysis
2.6. CRISPR
2.6.1. Design of the CRISPR Target Gene
2.6.2. Plasmid DNA Construct Design
2.6.3. Microinjection
2.6.4. Screening for CRISPR Knock-Out/in and Established Colonies
3. Results
3.1. Results
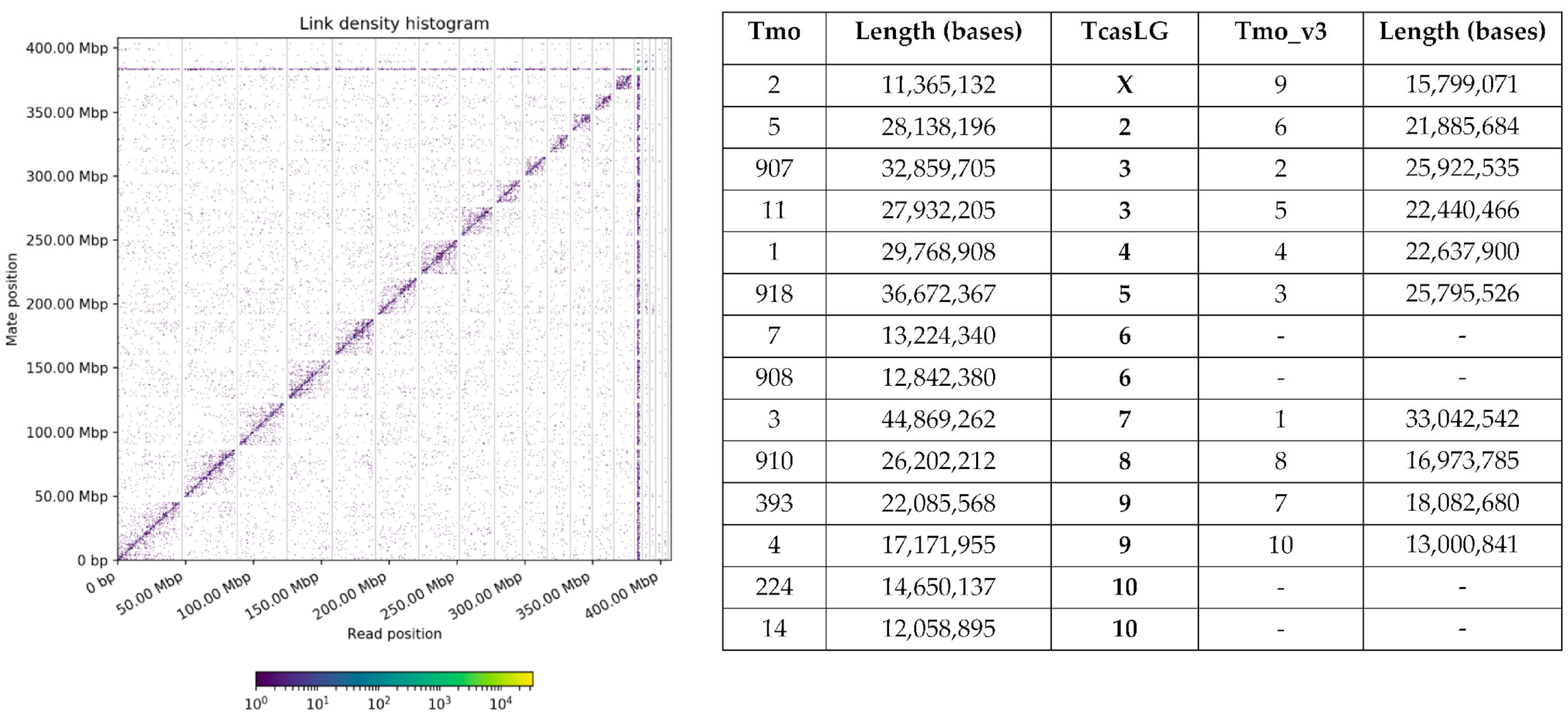
3.1.1. Genome Assembly of T. molitor
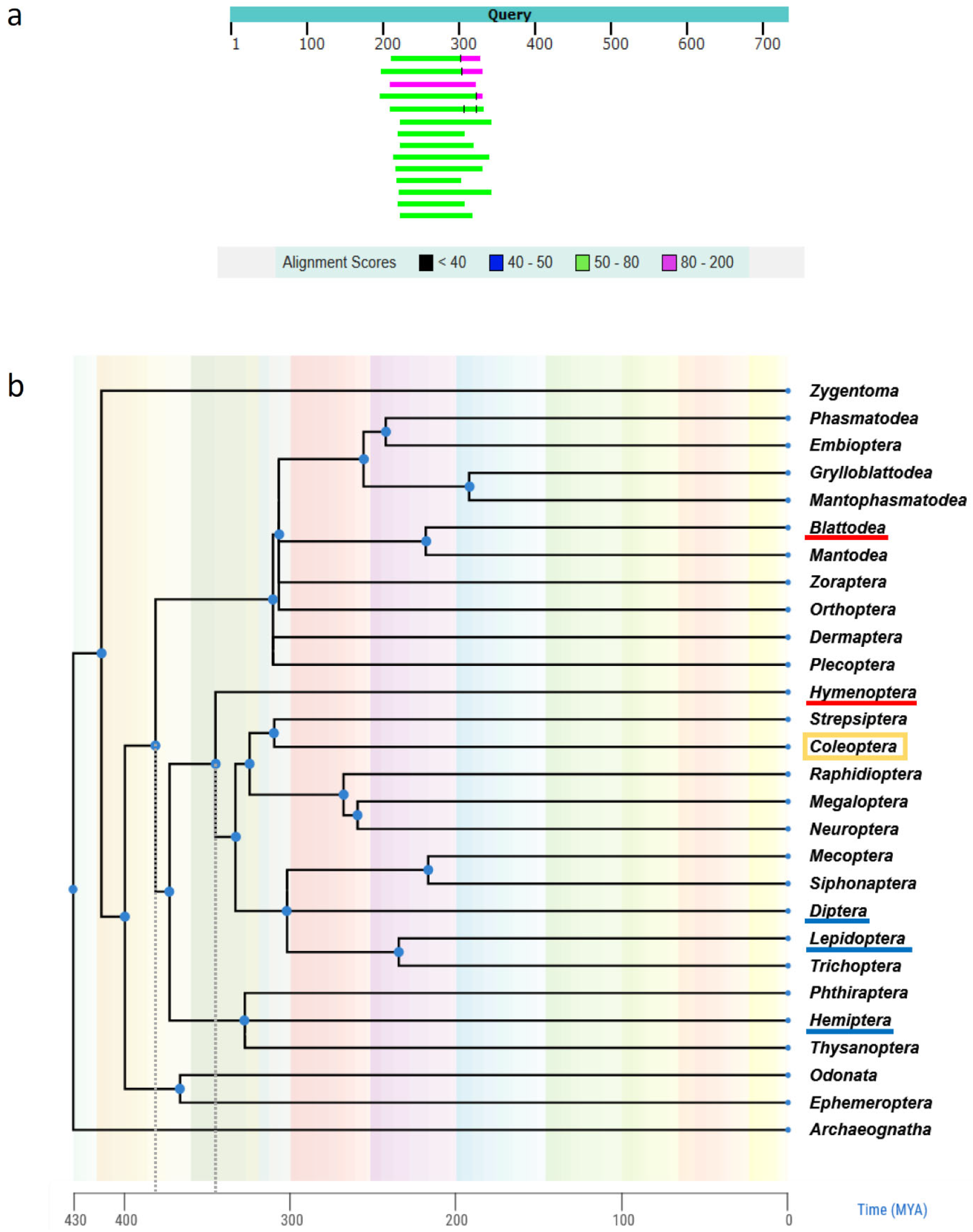
3.1.2. Comparison of T. molitor Genome Assemblies
3.2. Satellites in T. molitor
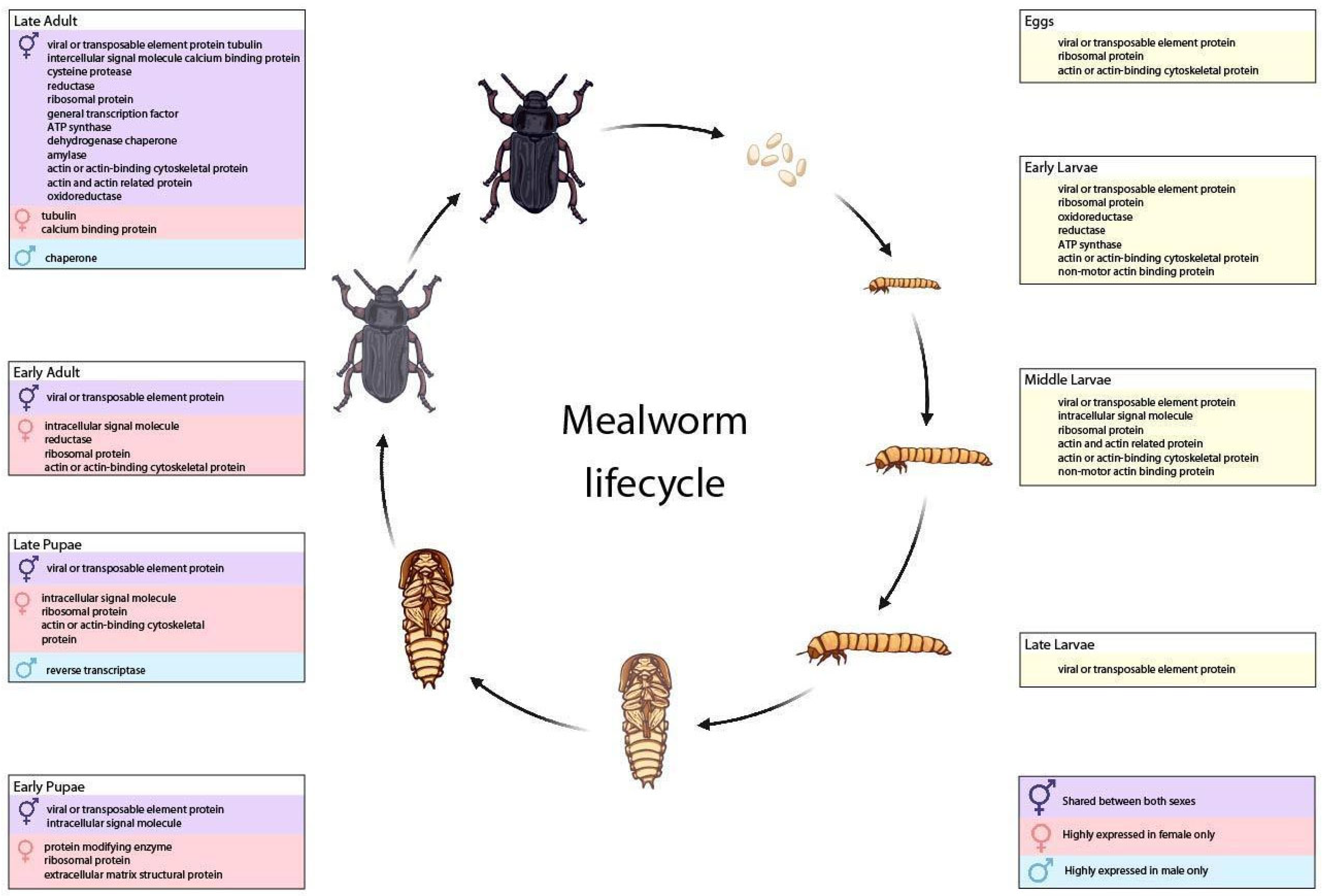
3.3. Transcripts from T. molitor Life Stages
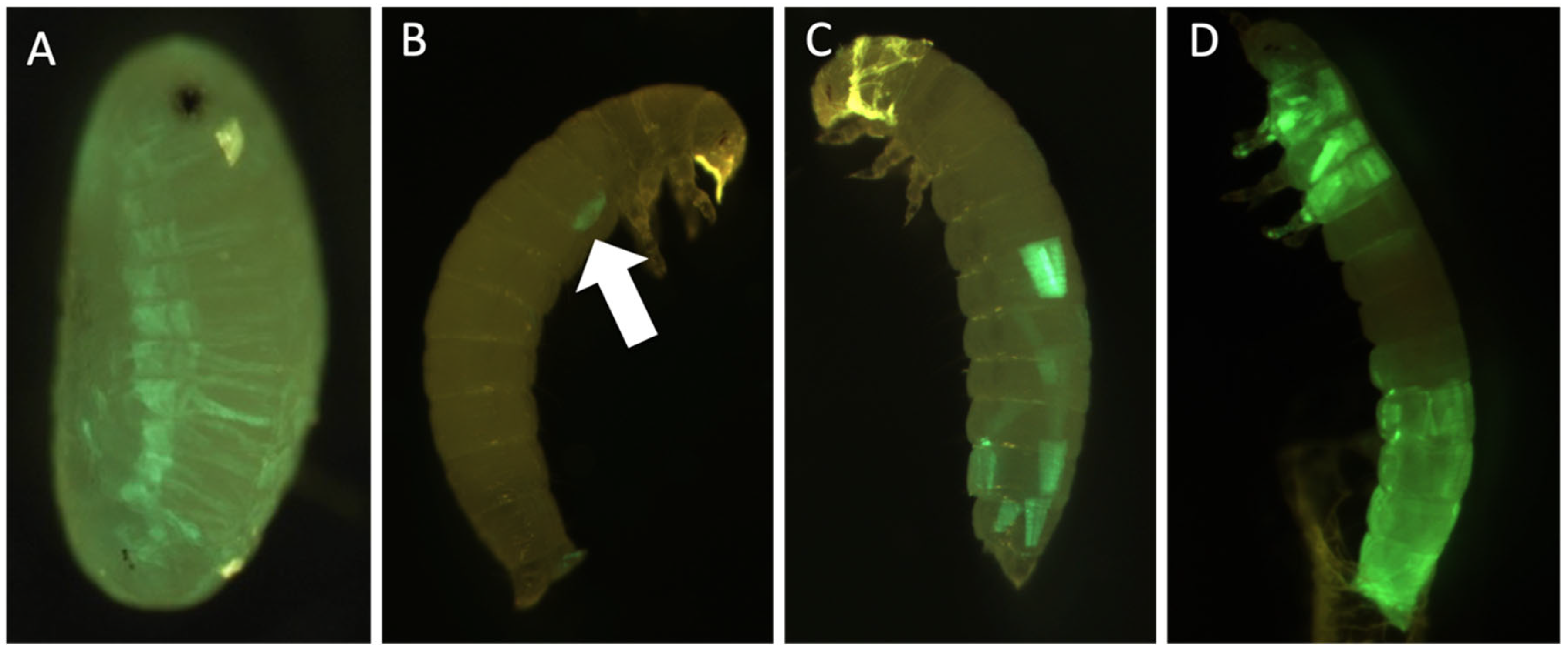
3.4. Genetic Engineering
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dossey, A.T.; Morales-Ramos, J.A.; Guadalupe Rojas, M. (Eds.) Insects as Sustainable Food Ingredients: Production, Processing and Food Applications; Academic Press: San Diego, CA, USA, 2016. [Google Scholar] [CrossRef]
- Oppert, B.; Perkin, L.C.; Lorenzen, M.; Dossey, A.T. Transcriptome analysis of life stages of the house cricket, Acheta domesticus, to improve insect crop production. Sci. Rep. 2020, 10, 3471. [Google Scholar] [CrossRef]
- Dossey, A.T.; Oppert, B.; Chu, F.-C.; Lorenzen, M.D.; Scheffler, B.; Simpson, S.; Koren, S.; Johnston, J.S.; Kataoka, K.; Ide, K. Genome and genetic engineering of the house cricket (Acheta domesticus): A resource for sustainable agriculture. Biomolecules 2023, 13, 589. [Google Scholar] [CrossRef]
- Dossey, A.T.; Tatum, J.T.; McGill, W.L. Modern Insect-Based Food Industry: Current Status, Insect Processing Technology, and Recommendations Moving Forward. In Insects as Sustainable Food Ingredients: Production, Processing and Food Applications; Dossey, A.T., Morales-Ramos, J.A., Guadalupe Rojas, M., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 113–152. [Google Scholar] [CrossRef]
- Williams, J.P.; Williams, J.R.; Kirabo, A.; Chester, D.; Peterson, M. Nutrient Content and Health Benefits of Insects. In Insects as Sustainable Food Ingredients: Production, Processing and Food Applications; Dossey, A.T., Morales-Ramos, J.A., Guadalupe Rojas, M., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 61–84. [Google Scholar] [CrossRef]
- Gahukar, R.T. Edible Insects Farming: Efficiency and Impact on Family Livelihood, Food Security, and Environment Compared with Livestock and Crops. In Insects as Sustainable Food Ingredients: Production, Processing and Food Applications; Dossey, A.T., Morales-Ramos, J.A., Guadalupe Rojas, M., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 61–84. [Google Scholar]
- van Huis, A.; Oonincx, D.G.A.B. The environmental sustainability of insects as food and feed. A review. Agron. Sustain. Dev. 2017, 37, 43. [Google Scholar] [CrossRef]
- Grau, T.; Vilcinskas, A.; Joop, G. Sustainable farming of the mealworm Tenebrio molitor for the production of food and feed. Z. Naturforsch C 2017, 72, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Salvia, R.; Scieuzo, C.; Schmitt, É.; Russo, A.; Falabella, P. Lipids from insects in cosmetics and for personal care products. Insects 2021, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Sönmez, E. The effect of different cold storage period on total lipid amount of Tenebrio molitor (coleoptera: Tenebrionidae) larvae. J. Anatol. Environ. Anim. Sci. 2021, 6, 449–455. [Google Scholar] [CrossRef]
- Losey, J.E.; Vaughan, M. The economic value of ecological services provided by insects. BioScience 2006, 56, 311–323. [Google Scholar] [CrossRef]
- Scudder, G.G. The Importance of Insects. In Insect Biodiversity; Foottit, R.G., Adler, P.H., Eds.; JohnWiley & Sons: Hoboken, NJ, USA, 2017; pp. 9–43. [Google Scholar]
- Noriega, J.A.; Hortal, J.; Azcárate, F.M.; Berg, M.P.; Bonada, N.; Briones, M.J.; Del Toro, I.; Goulson, D.; Ibanez, S.; Landis, D.A.; et al. Research trends in ecosystem services provided by insects. Basic Appl. Ecol. 2018, 26, 8–23. [Google Scholar] [CrossRef]
- Saunders, M.E.; Rader, R. Ecosystem Services of Insects. In Encyclopedia of Social Insects; Starr, C., Ed.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Donkersley, P.; Ashton, L.; Lamarre, G.P.A.; Segar, S. Global insect decline is the result of willful political failure: A battle plan for entomology. Ecol. Evol. 2022, 12, e9417. [Google Scholar] [CrossRef]
- Rubio, N.; Fish, K.; Trimmer, B.; Kaplan, D. Possibilities for engineered insect tissue as a food source. Front. Sustain. Food Syst. 2019, 3, 24. [Google Scholar] [CrossRef]
- Czekała, W. Concept of in-oil project based on bioconversion of by-products from food processing industry. J. Ecol. Eng. 2017, 18, 180–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Madau, F.; Arru, B.; Furesi, R.; Pulina, P. Insect farming for feed and food production from a circular business model perspective. Sustainability 2020, 12, 5418. [Google Scholar] [CrossRef]
- Moruzzo, R.; Riccioli, F.; Diaz, S.E.; Secci, C.; Poli, G.; Mancini, S. Mealworm (Tenebrio molitor): Potential and challenges to promote circular economy. Animals 2021, 11, 2568. [Google Scholar] [CrossRef] [PubMed]
- Smetana, S.; Spykman, R.; Heinz, V. Environmental aspects of insect mass production. J. Insects Food Feed. 2021, 7, 553–571. [Google Scholar] [CrossRef]
- Grigoropoulos, C.J.; Zerefos, S.; Tsangrassoulis, A.; Doulos, L.T. Lighting products as part of the circular economy and strategies that affect it. A literature overview. IOP Conf. Ser. Earth Environ. Sci. 2022, 1123, 012006. [Google Scholar] [CrossRef]
- Eriksson, T.; Andere, A.A.; Kelstrup, H.C.; Emery, V.; Picard, C. The yellow mealworm (Tenebrio molitor) genome: A resource for the emerging insects as food and feed industry. J. Insects Food Feed. 2020, 6, 445–455. [Google Scholar] [CrossRef]
- Eleftheriou, E.; Aury, J.M.; Vacherie, B.; Istace, B.; Belser, C.; Noel, B.; Moret, Y.; Rigaud, T.; Berro, F.; Gasparian, S.; et al. Chromosome-scale assembly of the yellow mealworm genome [version 3; peer review: 2 approved]. Open Res. Eur. 2022, 1, 94. [Google Scholar] [CrossRef]
- Kaur, S.; Stinson, S.A.; diCenzo, G.C. Whole genome assemblies of Zophobas morio and Tenebrio molitor. G3 Genes|Genomes|Genet. 2023, 13, jkad079. [Google Scholar] [CrossRef]
- Johnston, J.S.; Bernardini, A.; Hjelmen, C.E. Genome size estimation and quantitative cytogenetics in insects. Methods Mol. Biol. 2019, 1858, 15–26. [Google Scholar]
- Oppert, B.; Stoss, S.; Monk, A.; Smith, T. Optimized extraction of insect genomic DNA for long-read sequencing. Methods Protoc. 2019, 2, 89. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef]
- Oppert, B.; Muszewska, A.; Steczkiewicz, K.; Šatović-Vukšić, E.; Plohl, M.; Fabrick, J.A.; Vinokurov, K.S.; Koloniuk, I.; Johnston, J.S.; Smith, T.P.L.; et al. The genome of Rhyzopertha dominica (Fab.) (Coleoptera: Bostrichidae): Adaptation for success. Genes 2022, 13, 446. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 822. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol update for large-scale genome and gene function analysis with the Panther classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Hipp, R.D. SQLite. 2020. Available online: https://www.sqlite.org/index.html (accessed on 1 February 2023).
- Novák, P.; Robledillo, L.A.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.E.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Oppert, B.; Chu, F.-C.; Reyna, S.; Pinzi, S.; Adrianos, S.; Perkin, L.; Lorenzen, M. Effects of targeting eye color in Tenebrio molitor through RNA interference of tryptophan 2,3-dioxygenase (vermilion): Implications for insect farming. Arch. Insect Biochem. Physiol. 2019, 101, e21546. [Google Scholar] [CrossRef]
- Petitpierre, E.; Gatewood, J.M.; Schmid, C.W. Satellite DNA from the beetle Tenebrio molitor. Experientia 1988, 44, 498–499. [Google Scholar] [CrossRef]
- van Huis, A. Edible insects are the future? Proc. Nutr. Soc. 2016, 75, 294–305. [Google Scholar] [CrossRef]
- Martynov, A.G.; Elpidina, E.N.; Perkin, L.; Oppert, B. Functional analysis of C1 family cysteine peptidases in the larval gut of Tenebrio molitor and Tribolium castaneum. BMC Genomics 2015, 16, 75. [Google Scholar] [CrossRef]
- Šatović, E.; Cvitanić, M.T.; Plohl, M. Tools and databases for solving problems in detection and identification of repetitive DNA sequences. Period. Biol. 2020, 121–122, 7–14. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; MacAs, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Juan, C.; Gosálvez, J.; Mezzanotte, R.; Petitpierre, E. Cytological and biochemical characterization of the in situ endonuclease digestion of fixed Tenebrio molitor chromosomes. Chromosoma 1991, 100, 432–438. [Google Scholar] [CrossRef]
- Petitpierre, E.; Juan, C.; Pons, J.; Plohl, M.; Ugarkovic, D. Satellite DNA and Constitutive Heterochromatin in Tenebrionid Beetles. In Kew Chromosome Conference IV; Brandham, P.E., Bennet, M., Eds.; Royal Botanic Gardens: Richmond, UK, 1995; pp. 351–362. [Google Scholar]
- Davis, C.A.; Wyatt, G.R. Distribution and sequence homogeneity of an abundant satellite DNA in the beetle, Tenebrio molitor. Nucleic Acids Res. 1989, 17, 5579–5586. [Google Scholar] [CrossRef]
- Juan, C.; Petitpierre, E. C-banding and DNA content in seven species of Tenebrionidae (Coleoptera). Genome 1989, 32, 834–839. [Google Scholar] [CrossRef]
- Plohl, M.; Borstnik, B.; Lucijanić-Justić, V.; Ugarković, D. Evidence for random distribution of sequence variants in Tenebrio molitor satellite DNA. Genet. Res. 1992, 60, 7–13. [Google Scholar] [CrossRef]
- Masumoto, H.; Masukata, H.; Muro, Y.; Nozaki, N.; Okazaki, T.A. Human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 1989, 109, 1963–1973. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
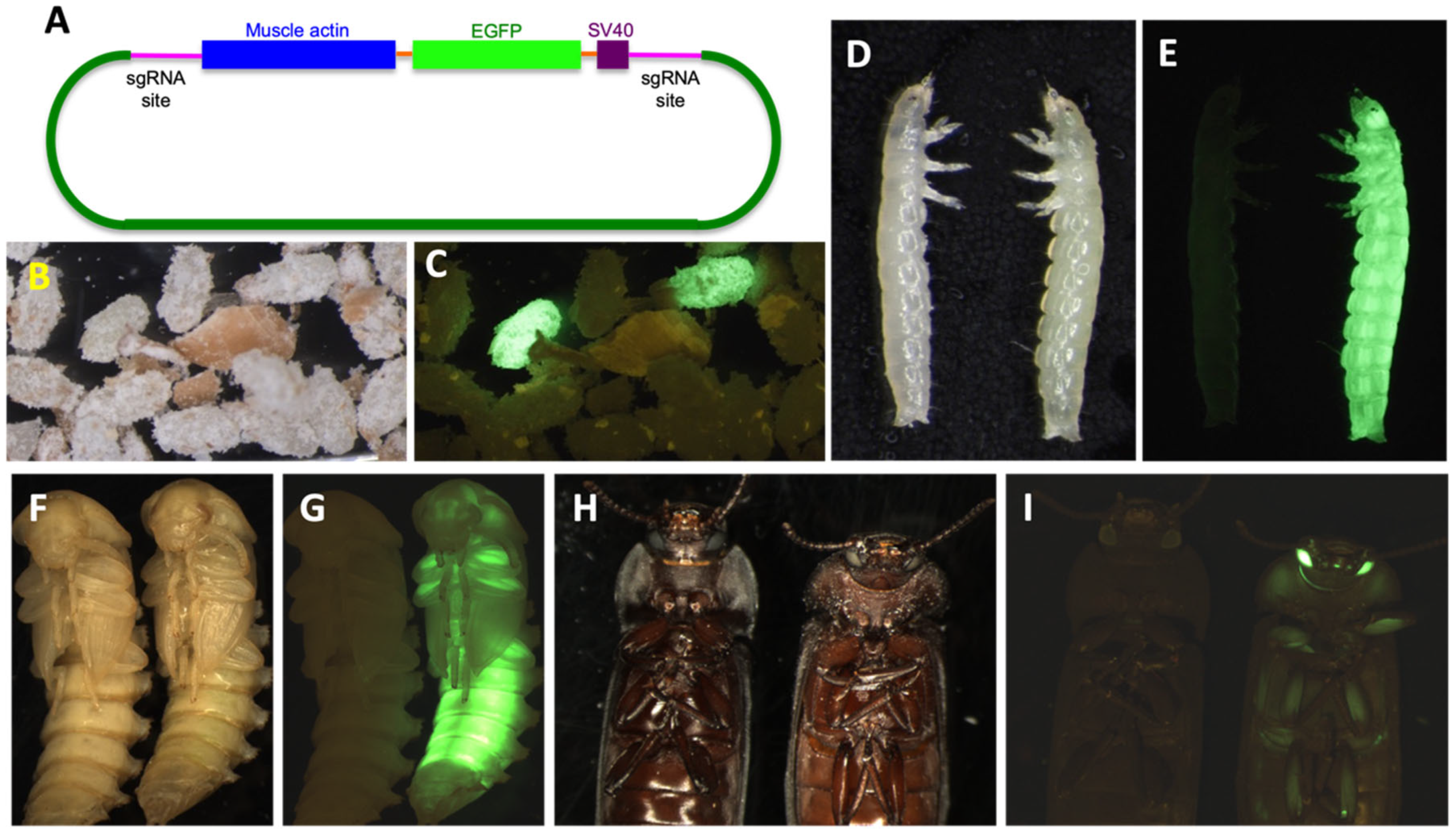{kind=link}
| Assembly | #Scaffolds | Total Bases | N50 | Longest Scaffold | BUSCO (%) |
|---|---|---|---|---|---|
| CANU | 7484 | 417,676,750 | 103,701 | 1,144,088 | 96.1 |
| Chicago/HiRise | 2364 | 423,052,750 | 2,013,304 | 9,264,513 | 96.2 |
| Hybrid | 1293 | 400,747,566 | 2,120,994 | 9,265,900 | 96.3 |
| Hi-C/HiRise | 1031 | 400,765,366 | 26,202,000 | 44,869,262 | 98.3 |
| Metrics | Tmo (This Study) | GCA_907166875.3 [23] | GCA_027725215.1 [24] |
|---|---|---|---|
| Total length (Mb) | 401 | 288 | 258 |
| Percent of genome | 53 | 38 | 33 |
| Tissue | single male pupa | single male pupa | head and legs |
| Sequencing technology | PacBio RSII P6 | Illumina PCR-free, PromethION | Oxford Nanopore MinION; Illumina NovaSeq |
| Predicted #genes | 35,281 | 21,418 | 19,830 |
| Number of scaffolds | 1031 | 111 | 1986 |
| Scaffold N50 (Mb) | 26.2 | 21.9 | 20.8 |
| Scaffold L50 | 7 | 6 | 6 |
| Scaffolding technology | Dovetail Chicago/Hi-C | Dovetail Hi-C | n/a |
| satDNA | Assembly-Free TAREAN Analysis | Genome Assembly | |||||
|---|---|---|---|---|---|---|---|
| Monomer Length (bp) | Abundance (%) | Number of Monomers | Number of Scaffolds Occupied | Average Monomer Identity (%) | bp Occupied | % of the Assembly | |
| TmSat1 | 142 | 26.50 | 11654 | 36 | 98.1 | 1,654,228 | 0.413 |
| TmSat2 | 180 | 0.45 | 784 | 6 | 96.2 | 141,131 | 0.035 |
| TmSat3 | 325 | 0.38 | 2360 | 19 | 98.6 | 765,823 | 0.191 |
| TmSat4 | 245 | 0.26 | 637 | 13 | 87.2 | 155,006 | 0.038 |
| TmSat5 | 364 | 0.15 | 781 | 33 | 90.4 | 282,105 | 0.070 |
| TmSat6 | 227 | 0.11 | 157 | 10 | 92.1 | 35,682 | 0.009 |
| TmSat7 | 189 | 0.08 | 89 | 6 | 97.0 | 16,800 | 0.004 |
| TmSat8 | 735 | 0.05 | 127 | 4 | 93.9 | 92,660 | 0.023 |
| TmSat9 | 108 | 0.02 | 192 | 1 | 83.8 | 20,750 | 0.005 |
| TmSat10 | 150 | 0.01 | 28 | 1 | 92.6 | 4,210 | 0.001 |
| TmSat11 | 93 | 0.01 | 6 | 1 | 86.8 | 537 | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oppert, B.; Dossey, A.T.; Chu, F.-C.; Šatović-Vukšić, E.; Plohl, M.; Smith, T.P.L.; Koren, S.; Olmstead, M.L.; Leierer, D.; Ragan, G.; et al. The Genome of the Yellow Mealworm, Tenebrio molitor: It’s Bigger Than You Think. Genes 2023, 14, 2209. https://doi.org/10.3390/genes14122209
Oppert B, Dossey AT, Chu F-C, Šatović-Vukšić E, Plohl M, Smith TPL, Koren S, Olmstead ML, Leierer D, Ragan G, et al. The Genome of the Yellow Mealworm, Tenebrio molitor: It’s Bigger Than You Think. Genes. 2023; 14(12):2209. https://doi.org/10.3390/genes14122209
Chicago/Turabian StyleOppert, Brenda, Aaron T. Dossey, Fu-Chyun Chu, Eva Šatović-Vukšić, Miroslav Plohl, Timothy P. L. Smith, Sergey Koren, Morgan L. Olmstead, Dewey Leierer, Gail Ragan, and et al. 2023. "The Genome of the Yellow Mealworm, Tenebrio molitor: It’s Bigger Than You Think" Genes 14, no. 12: 2209. https://doi.org/10.3390/genes14122209
APA StyleOppert, B., Dossey, A. T., Chu, F.-C., Šatović-Vukšić, E., Plohl, M., Smith, T. P. L., Koren, S., Olmstead, M. L., Leierer, D., Ragan, G., & Johnston, J. S. (2023). The Genome of the Yellow Mealworm, Tenebrio molitor: It’s Bigger Than You Think. Genes, 14(12), 2209. https://doi.org/10.3390/genes14122209