
Comprehensive Identification of Mitochondrial Pseudogenes (NUMTs) in the Human Telomere-to-Telomere Reference Genome


Abstract
:1. Introduction
2. Data and Methods
2.1. Preparing the Human Pan-Mitogenome (HPMT)
2.2. BLAST and Filtering
2.3. Alignment of Short Segments
2.4. Eliminating Mitochondrial Copy Numbers in HEK293 Cell Lines
2.5. ATAC-Seq Data Analysis
2.6. Statistics and Visualization
3. Results
3.1. Human Pan-Mitogenome (HPMT)
3.2. More NUMTs Were Identified Using HPMT
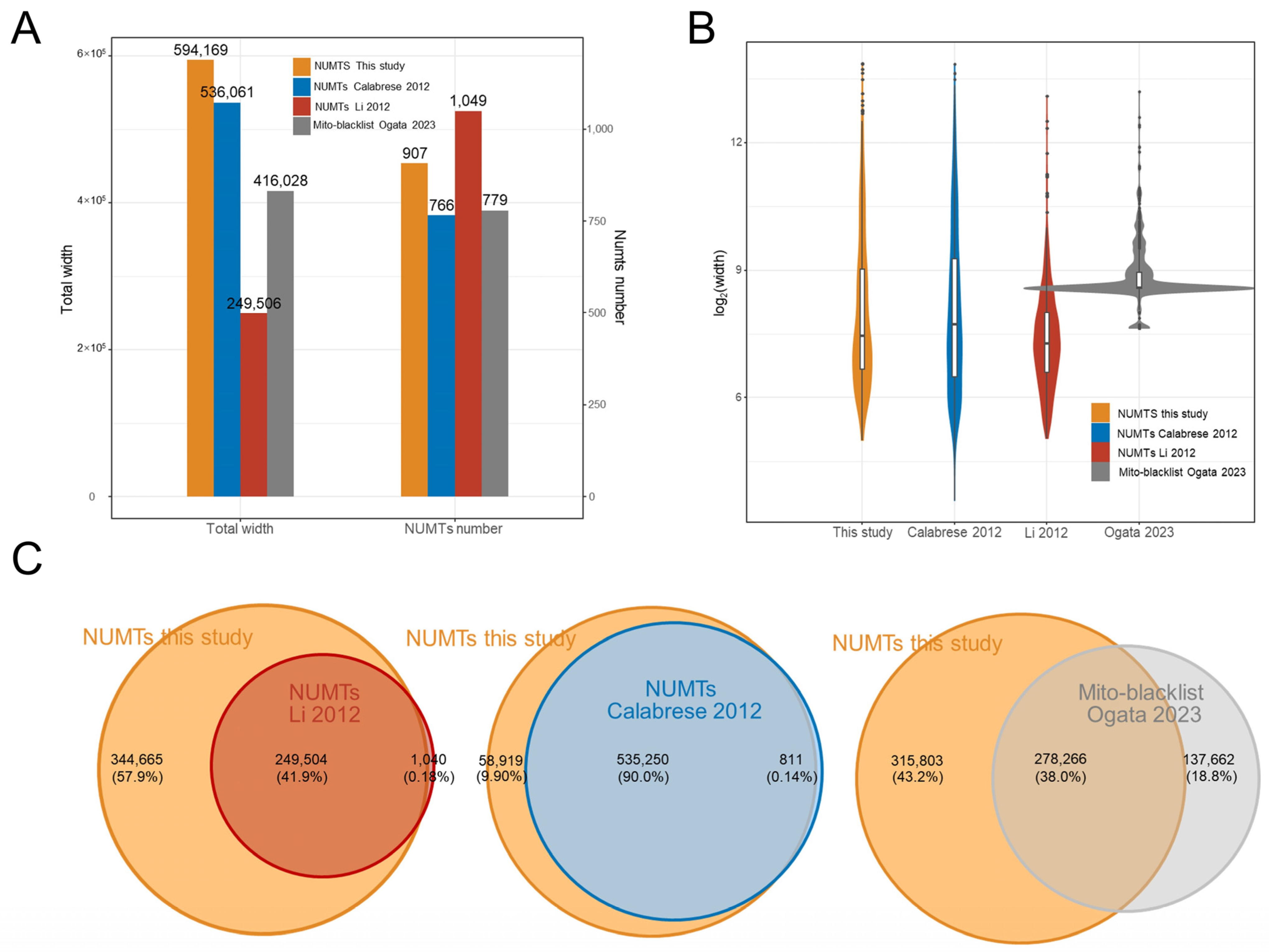
3.3. Comparison with the Known NUMTs Compilation
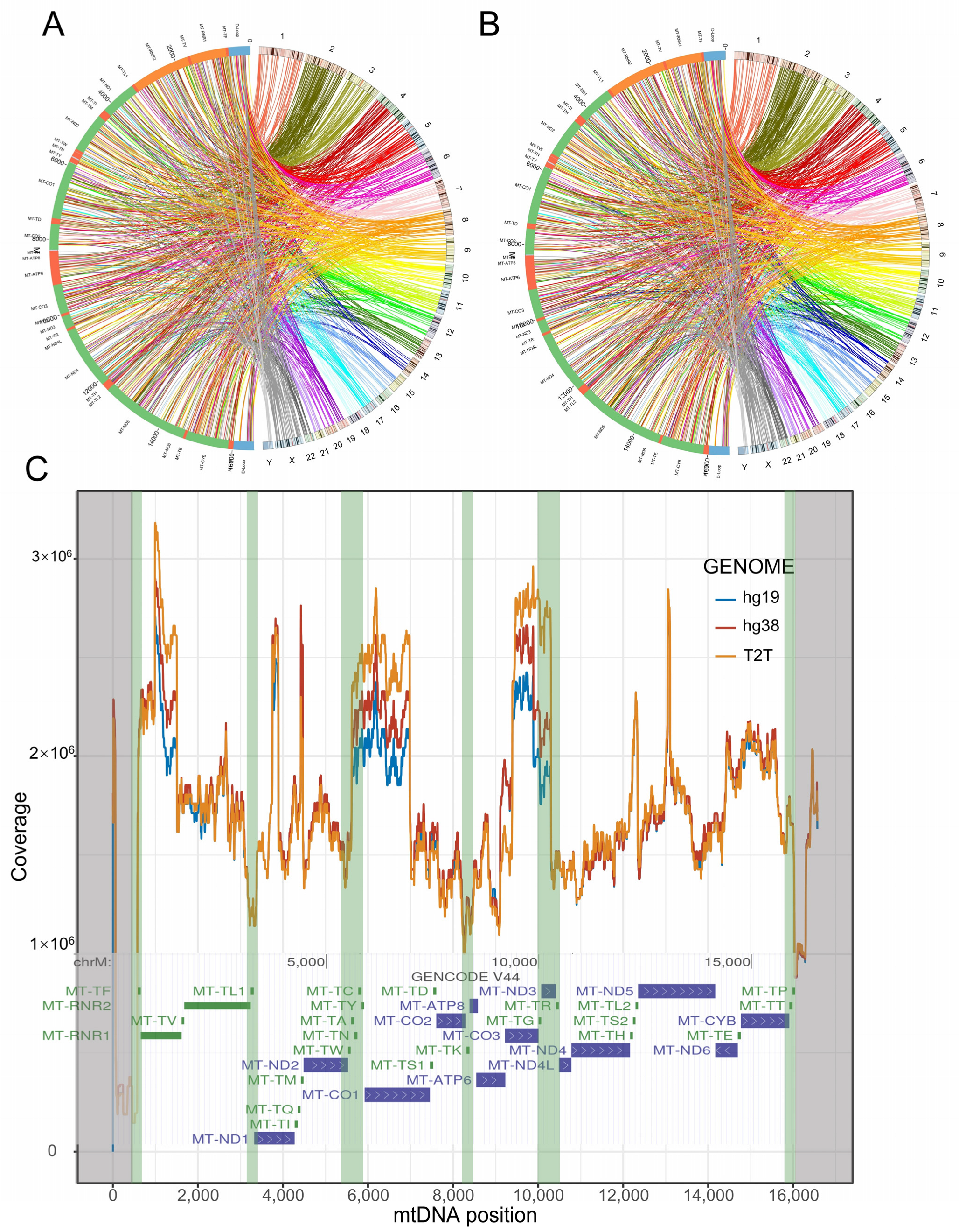
3.4. Distribution of NUMTs in the Human Reference Genome
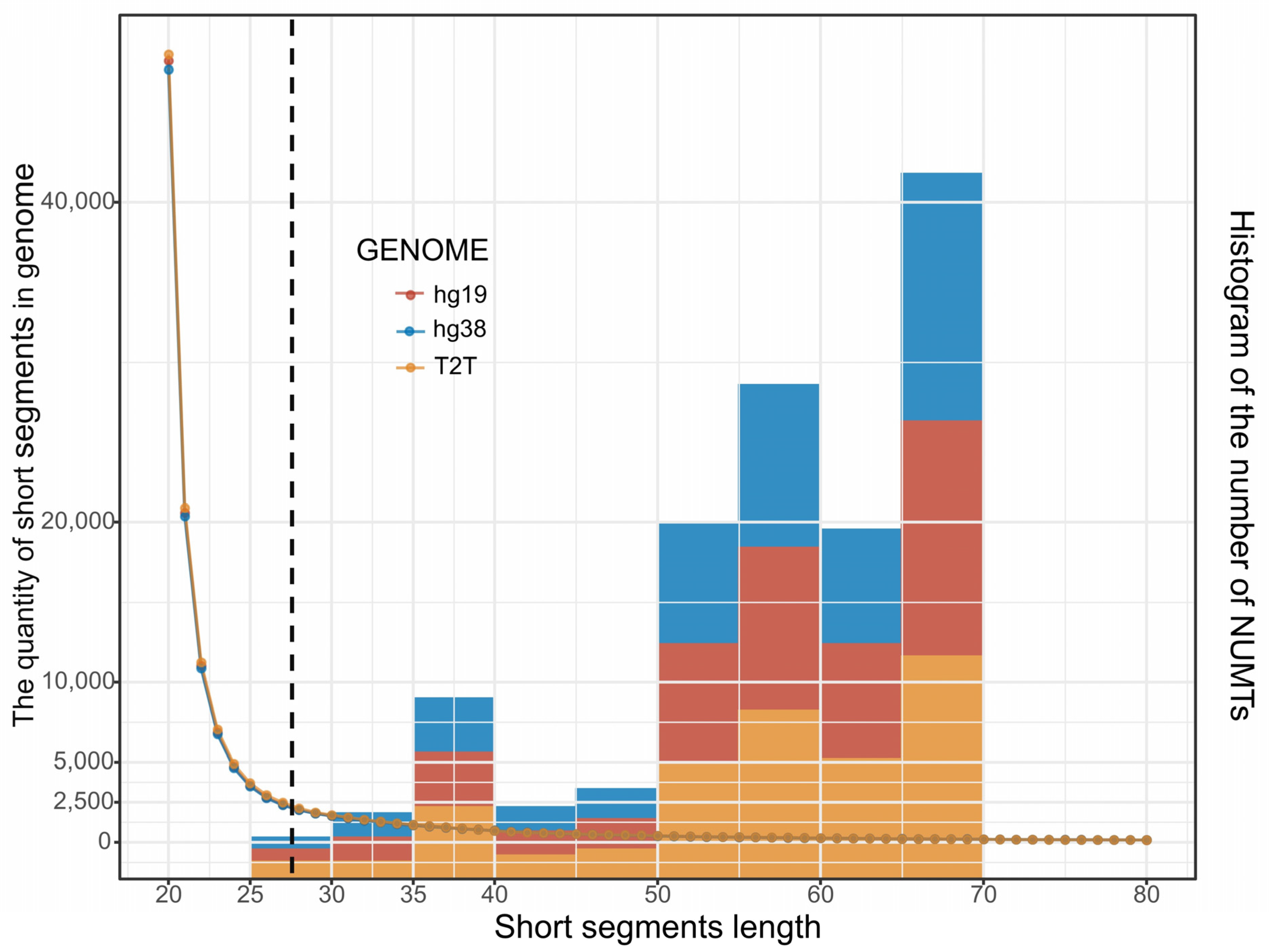
3.5. mtDNA-like Short Segments within the Nuclear Genome
3.6. The Impact of NUMTs on ATAC-Seq Appears to Be Limited
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woischnik, M.; Moraes, C.T. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 2002, 12, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.V.; Yuhki, N.; Masuda, R.; Modi, W.; O’Brien, S.J. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 1994, 39, 174–190. [Google Scholar] [CrossRef]
- Bravi, C.M.; Parson, W.; Bandelt, H.-J. Numts Revisited. In Human Mitochondrial DNA and the Evolution of Homo Sapiens; Bandelt, H.-J., Macaulay, V., Richards, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 31–46. [Google Scholar] [CrossRef]
- Woerner, A.E.; Cihlar, J.C.; Smart, U.; Budowle, B. Numt identification and removal with RtN! Bioinformatics 2020, 36, 5115–5116. [Google Scholar] [CrossRef]
- Wei, W.; Schon, K.R.; Elgar, G.; Orioli, A.; Tanguy, M.; Giess, A.; Tischkowitz, M.; Caulfield, M.J.; Chinnery, P.F. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 2022, 611, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.M.; Balacco, D.L.; Preste, R.; Diroma, M.A.; Forino, R.; Ventura, M.; Attimonelli, M. NumtS colonization in mammalian genomes. Sci. Rep. 2017, 7, 16357. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schroeder, R.; Ko, A.; Stoneking, M. Fidelity of capture-enrichment for mtDNA genome sequencing: Influence of NUMTs. Nucleic Acids Res. 2012, 40, e137. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.M.; Simone, D.; Attimonelli, M. Primates and mouse NumtS in the UCSC Genome Browser. BMC Bioinform. 2012, 13 (Suppl. S4), S15. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef]
- Maude, H.; Davidson, M.; Charitakis, N.; Diaz, L.; Bowers, W.H.T.; Gradovich, E.; Andrew, T.; Huntley, D. NUMT Confounding Biases Mitochondrial Heteroplasmy Calls in Favor of the Reference Allele. Front. Cell Dev. Biol. 2019, 7, 201. [Google Scholar] [CrossRef]
- Montefiori, L.; Hernandez, L.; Zhang, Z.; Gilad, Y.; Ober, C.; Crawford, G.; Nobrega, M.; Jo Sakabe, N. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Sci. Rep. 2017, 7, 2451. [Google Scholar] [CrossRef]
- Marshall, C.; Parson, W. Interpreting NUMTs in forensic genetics: Seeing the forest for the trees. Forensic Sci. Int. Genet. 2021, 53, 102497. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]
- Luo, J.; Shen, S.; Xia, J.; Wang, J.; Gu, Z. Mitochondria as the Essence of Yang Qi in the Human Body. Phenomics 2022, 2, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Liu, V.; Muus, C.; Praktiknjo, S.D.; Nitsch, L.; Kautz, P.; Sandor, K.; Yin, Y.; Gutierrez, J.C.; Pelka, K.; et al. Mitochondrial single-cell ATAC-seq for high-throughput multi-omic detection of mitochondrial genotypes and chromatin accessibility. Nat. Protoc. 2023, 18, 1416–1440. [Google Scholar] [CrossRef]
- Sturk-Andreaggi, K.; Ring, J.D.; Ameur, A.; Gyllensten, U.; Bodner, M.; Parson, W.; Marshall, C.; Allen, M. The Value of Whole-Genome Sequencing for Mitochondrial DNA Population Studies: Strategies and Criteria for Extracting High-Quality Mitogenome Haplotypes. Int. J. Mol. Sci. 2022, 23, 2244. [Google Scholar] [CrossRef] [PubMed]
- Ring, J.D.; Sturk-Andreaggi, K.; Alyse Peck, M.; Marshall, C. Bioinformatic removal of NUMT-associated variants in mitotiling next-generation sequencing data from whole blood samples. Electrophoresis 2018, 39, 2785–2797. [Google Scholar] [CrossRef]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.-C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Wei, W.; Rahman, S.; Chinnery, P.F. Biparental inheritance of mitochondrial DNA revisited. Nat. Rev. Genet. 2021, 22, 477–478. [Google Scholar] [CrossRef]
- Lutz-Bonengel, S.; Niederstatter, H.; Naue, J.; Koziel, R.; Yang, F.; Sanger, T.; Huber, G.; Berger, C.; Pflugradt, R.; Strobl, C.; et al. Evidence for multi-copy Mega-NUMTs in the human genome. Nucleic Acids Res. 2021, 49, 1517–1531. [Google Scholar] [CrossRef]
- Balciuniene, J.; Balciunas, D. A Nuclear mtDNA Concatemer (Mega-NUMT) Could Mimic Paternal Inheritance of Mitochondrial Genome. Front. Genet. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.; Gunbin, K.; Peshkin, L.; Annis, S.; Fleischmann, Z.; Franco, M.; Kraytsberg, Y.; Markuzon, N.; Ackermann, R.; Khrapko, K. Mitochondrial Pseudogenes Suggest Repeated Inter-Species Hybridization among Direct Human Ancestors. Genes. 2022, 13, 810. [Google Scholar] [CrossRef] [PubMed]
- Bucking, R.; Cox, M.P.; Hudjashov, G.; Saag, L.; Sudoyo, H.; Stoneking, M. Archaic mitochondrial DNA inserts in modern day nuclear genomes. BMC Genom. 2019, 20, 1017. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Graur, D. A comparative analysis of numt evolution in human and chimpanzee. Mol. Biol. Evol. 2007, 24, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Dayama, G.; Zhou, W.; Prado-Martinez, J.; Marques-Bonet, T.; Mills, R.E. Characterization of nuclear mitochondrial insertions in the whole genomes of primates. NAR Genom. Bioinform. 2020, 2, lqaa089. [Google Scholar] [CrossRef] [PubMed]
- Gherman, A.; Chen, P.E.; Teslovich, T.M.; Stankiewicz, P.; Withers, M.; Kashuk, C.S.; Chakravarti, A.; Lupski, J.R.; Cutler, D.J.; Katsanis, N. Population Bottlenecks as a Potential Major Shaping Force of Human Genome Architecture. PLoS Genet. 2007, 3, e119. [Google Scholar] [CrossRef]
- Gunbin, K.; Peshkin, L.; Popadin, K.; Annis, S.; Ackermann, R.; Khrapko, K. Integration of mtDNA pseudogenes into the nuclear genome coincides with speciation of the human genus. A hypothesis. Mitochondrion 2017, 34, 20–23. [Google Scholar] [CrossRef]
- Dayama, G.; Emery, S.B.; Kidd, J.M.; Mills, R.E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 2014, 42, 12640–12649. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Horai, S.; Hayasaka, K.; Kondo, R.; Tsugane, K.; Takahata, N. Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs. Proc. Natl. Acad. Sci. USA 1995, 92, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Ogata, J.D.; Mu, W.; Davis, E.S.; Xue, B.; Harrell, J.C.; Sheffield, N.C.; Phanstiel, D.H.; Love, M.I.; Dozmorov, M.G. excluderanges: Exclusion sets for T2T-CHM13, GRCm39, and other genome assemblies. Bioinformatics 2023, 39, btad198. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, X.; Chen, H.; Tan, X.; Yang, Z.; Deng, L.; Wang, B.; Kong, S.; Li, S.; Cui, Y.; et al. A pangenome reference of 36 Chinese populations. Nature 2023, 619, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstatter, A.; Forer, L.; Specht, G.; Bandelt, H.J.; Kronenberg, F.; Salas, A.; Schonherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef]
- Duguay, B.A.; Smiley, J.R. Mitochondrial nucleases ENDOG and EXOG participate in mitochondrial DNA depletion initiated by herpes simplex virus 1 UL12.5. J. Virol. 2013, 87, 11787–11797. [Google Scholar] [CrossRef]
- Spadafora, D.; Kozhukhar, N.; Chouljenko, V.N.; Kousoulas, K.G.; Alexeyev, M.F. Methods for Efficient Elimination of Mitochondrial DNA from Cultured Cells. PLoS ONE 2016, 11, e0154684. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinform. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 November 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Lutz-Bonengel, S.; Parson, W. No further evidence for paternal leakage of mitochondrial DNA in humans yet. Proc. Natl. Acad. Sci. USA 2019, 116, 1821–1822. [Google Scholar] [CrossRef]
- Hackl, T.; Laurenceau, R.; Ankenbrand, M.J.; Bliem, C.; Cariani, Z.; Thomas, E.; Dooley, K.D.; Arellano, A.A.; Hogle, S.L.; Berube, P.; et al. Novel integrative elements and genomic plasticity in ocean ecosystems. Cell 2023, 186, 47–62.e16. [Google Scholar] [CrossRef]
- Zhang, R.; Nakahira, K.; Guo, X.; Choi, A.M.; Gu, Z. Very Short Mitochondrial DNA Fragments and Heteroplasmy in Human Plasma. Sci. Rep. 2016, 6, 36097. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Previous Studies | NUMTs | Total Length | Min | Median | Mean | Max | E-Value_Max | Iden_Min |
| hg19_2012_Calabrese | 766 | 536,061 | 12 | 212 | 700 | 14,835 | 10 | 63.52 |
| hg19_2012_Li | 1049 | 249,506 | 33 | 155 | 238 | 8795 | 0.00005 | 78.11 |
| This Study | NUMTs | Total Length | Min | Median | Mean | Max | E-Value_Max | Iden_Min |
| hg19 | 907 | 594,169 | 32 | 176 | 655 | 14,855 | 0.0001 | 63.89 |
| hg38 | 908 | 596,516 | 32 | 176 | 657 | 14,855 | 0.0001 | 63.89 |
| T2T-CHR13 | 958 | 631,156 | 28 | 194 | 659 | 14,855 | 0.0001 | 63.37 |
| Total Segments | Unique Segments | Reference | Mapped Pos | Total Length | Min | Median | Mean | Max | >30 Pos |
|---|---|---|---|---|---|---|---|---|---|
| 854,733,854 | 423,658 | hg19 | 200,968 | 4,172,251 | 20 | 20 | 21 | 5862 | 1808 |
| hg38 | 199,556 | 4,142,766 | 20 | 20 | 21 | 5862 | 1802 | ||
| T2T-CHR13 | 202,859 | 4,207,476 | 20 | 20 | 21 | 3420 | 1872 |
| Reference | Number | Total Length | Min | Median | Mean | Max |
|---|---|---|---|---|---|---|
| hg19 | 1978 | 901,808 | 20 | 194 | 456 | 15,010 |
| hg38 | 1964 | 903,362 | 20 | 198 | 460 | 15,010 |
| T2T-CHR13 | 1595 | 867,093 | 20 | 301 | 544 | 15,010 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, Y.; He, C.; Lin, D.; Gu, Z.; Pu, W. Comprehensive Identification of Mitochondrial Pseudogenes (NUMTs) in the Human Telomere-to-Telomere Reference Genome. Genes 2023, 14, 2092. https://doi.org/10.3390/genes14112092
Tao Y, He C, Lin D, Gu Z, Pu W. Comprehensive Identification of Mitochondrial Pseudogenes (NUMTs) in the Human Telomere-to-Telomere Reference Genome. Genes. 2023; 14(11):2092. https://doi.org/10.3390/genes14112092
Chicago/Turabian StyleTao, Yichen, Chengpeng He, Deng Lin, Zhenglong Gu, and Weilin Pu. 2023. "Comprehensive Identification of Mitochondrial Pseudogenes (NUMTs) in the Human Telomere-to-Telomere Reference Genome" Genes 14, no. 11: 2092. https://doi.org/10.3390/genes14112092
APA StyleTao, Y., He, C., Lin, D., Gu, Z., & Pu, W. (2023). Comprehensive Identification of Mitochondrial Pseudogenes (NUMTs) in the Human Telomere-to-Telomere Reference Genome. Genes, 14(11), 2092. https://doi.org/10.3390/genes14112092