

NGS Custom Panel Implementation in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital
 , , and
, , and
Abstract
1. Introduction
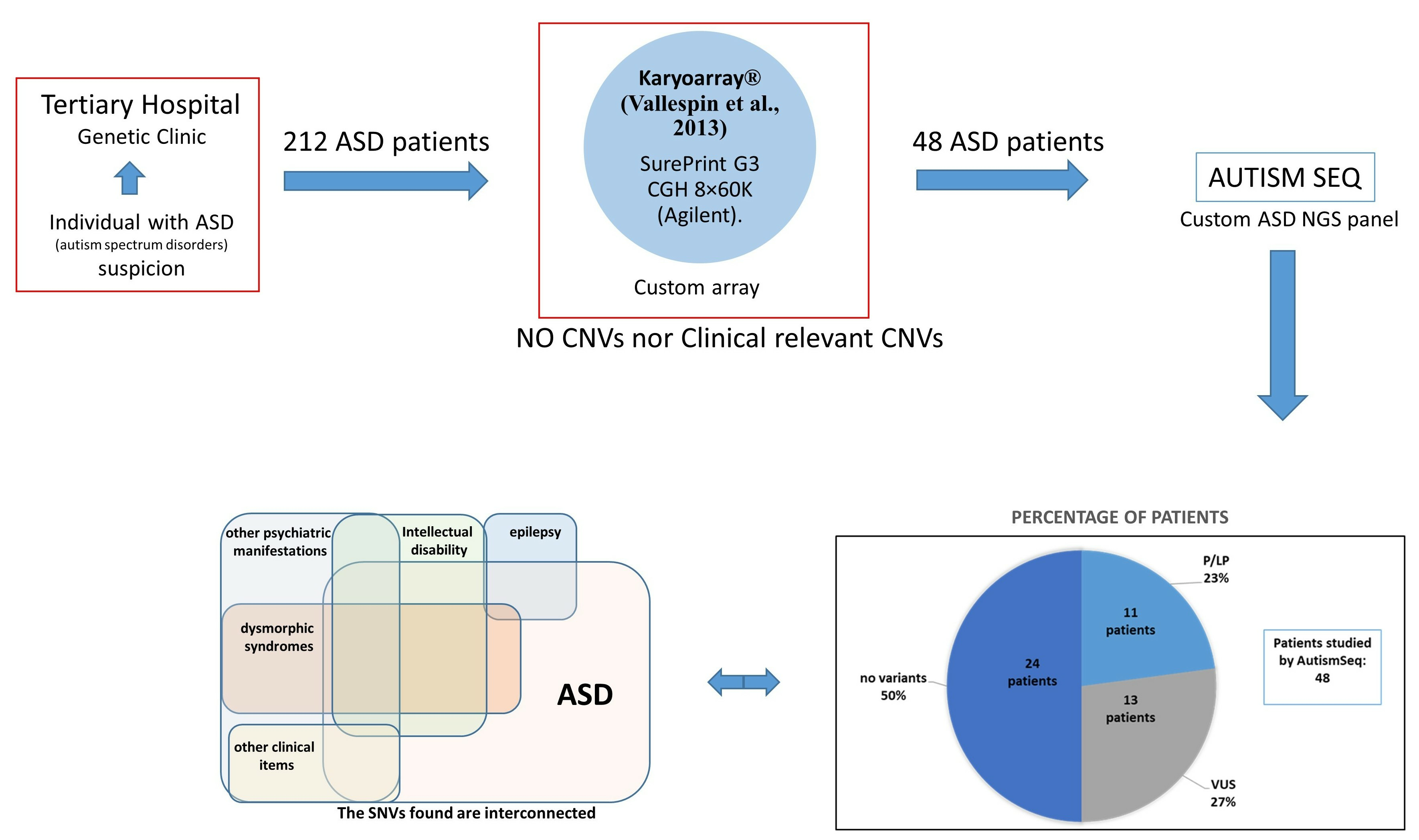
2. Materials and Methods
2.1. Genetic Studies
2.1.1. Karyotyping and Fluorescence In Situ Hybridization (FISH) Analysis
2.1.2. Fragile-X Syndrome Analysis
2.1.3. Multiplex Ligation-Dependent Probe Amplification (MLPA) Analysis
2.1.4. Chromosomal Microarray (CMA) Analysis
2.1.5. Next Generation Sequencing (NGS) Custom Panel Design
2.1.6. Next Generation Sequencing (NGS) Custom Panel Analysis
2.2. Study Limitations
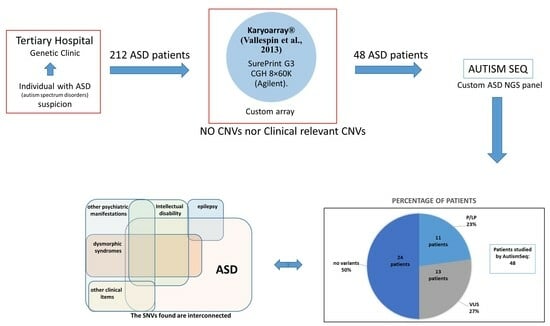
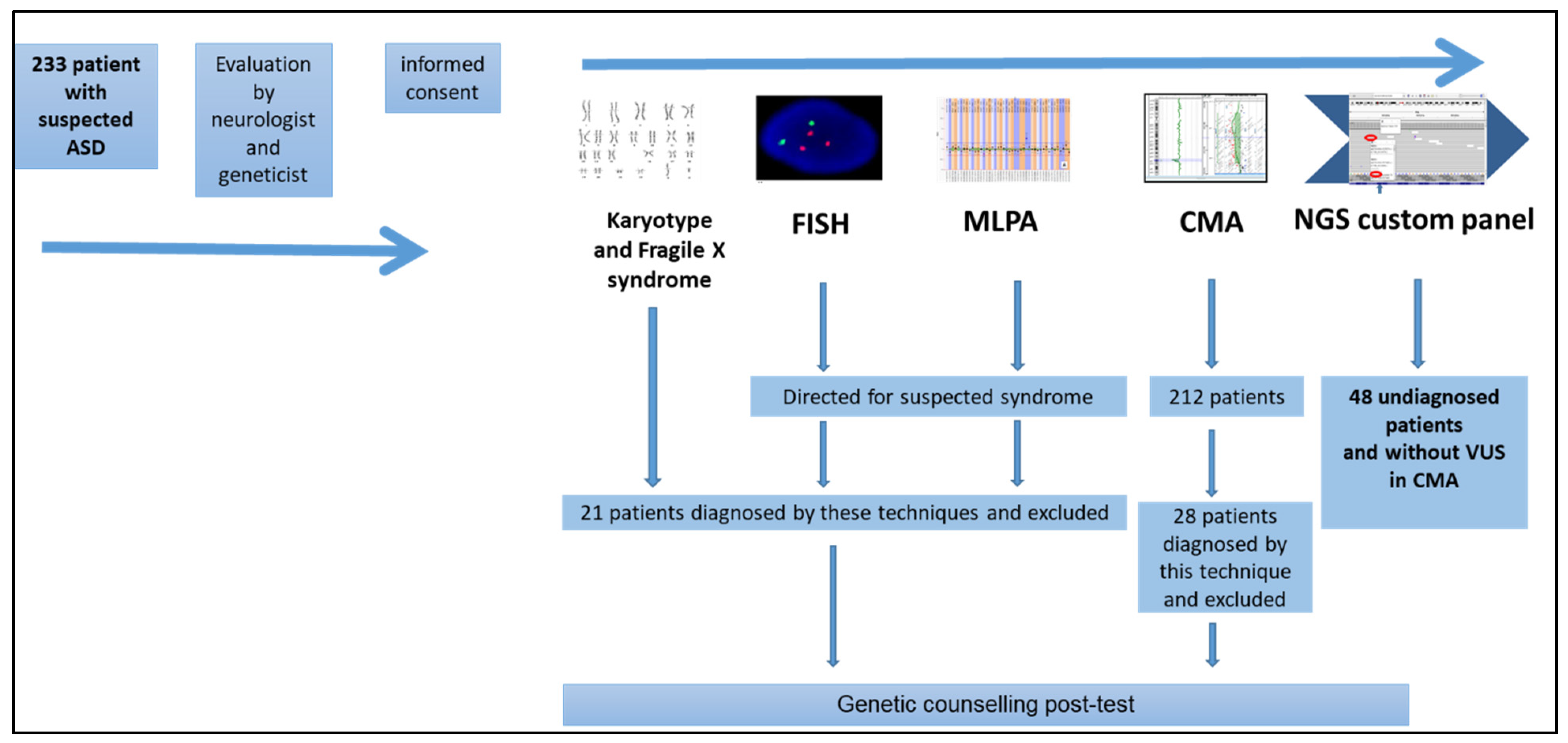
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Márquez, C.M.; Albores, G.L. Autistic spectrum disorders: Diagnostic and therapeutic challenges in Mexico. Salud Ment. 2011, 34, 435–441. [Google Scholar]
- Rosen, N.E.; Lord, C.; Volkmar, F.R. The Diagnosis of Autism: From Kanner to DSM-III to DSM-5 and beyond. J. Autism Dev. Disord. 2021, 51, 4253–4270. [Google Scholar] [CrossRef]
- McPartland, J.C.; Reichow, B.; Volkmar, F.R. Sensitivity and Specificity of Proposed DSM-5 Diagnostic Criteria for Autism Spectrum Disorder. J. Am. Acad. Child Adolesc. Psychiatr. 2012, 51, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Sevilla, F.; Bermúdez, E.; Sánchez, C. Estimated prevalence of autism spectrum disorders in the Canary Islands. An. Pediatr. 2013, 79, 352–359. [Google Scholar]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 20, 385. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef]
- Gazzellone, M.J.; Zhou, X.; Lionel, A.C.; Uddin, M.; Thiruvahindrapuram, B.; Liang, S. Copy number variation in Han Chinese individuals with autism spectrum disorder. J. Neurodev. Disord. 2014, 6, 34. [Google Scholar] [CrossRef]
- Bhandari, R.; Paliwal, J.K.; Kuhad, A. Neuropsychopathology of Autism Spectrum Disorder: Complex Interplay of Genetic, Epigenetic, and Environmental Factors. Adv. Neurobiol. 2020, 24, 97–141. [Google Scholar] [PubMed]
- Hodges, H.; Fealko, C.; Soares, N. Autism spectrum disorder: Definition, epidemiology, causes, and clinical evaluation. Transl. Pediatr. 2020, 9, S55–S65. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Ungar, J. Next Generation Sequencing and Health Technology Assessment in Autism Spectrum Disorder. J. Can. Acad. Child Adolesc. Psychiatry 2015, 24, 123–127. [Google Scholar]
- Fombonne, E. Epidemiological surveys of autism and other pervasive developmental disorders: An update. J. Autism Dev. Disord. 2003, 33, 365–382. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Sertié, A.L. Autism spectrum disorders: An updated guide for genetic counseling. Einstein 2017, 15, 233–238. [Google Scholar] [CrossRef]
- Peixoto, S.; Melo, J.B.; Ferrão, J.; Pires, L.M.; Lavoura, N.; Pinto, M.; Oliveira, G.; Carreira, I.M. MLPA analysis in a cohort of patients with autism. Mol. Cytogenet. 2017, 4, 10–12. [Google Scholar] [CrossRef][Green Version]
- Sherer, S.W.; Dawson, G. Risk factors for autism: Translating genomic discoveries into diagnostics. Hum. Genet. 2011, 130, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Kanduri, C.; Kantojarvi, K.; Salo, P.M.; Vanhala, R.; Buck, G.; Blancher, C.; Lahdesmaki, H.; Irma, J. The Landscape of Copy Number Variations in Finnish Families with Autism Spectrum Disorder. Autism Res. 2016, 9, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Mora, M.I.; Escalona, R.C.; Navarro, O.P.; Madrigal, I.; Quintela, I.; Amigo, J.; Martinez-Elurbe, D.; Linder-Lucht, M.; Aznar Lain, G.; Carracedo, A. Comprehensive molecular testing in patients with a high functioning autism spectrum disorder. Mutat. Res. 2016, 784, 46–52. [Google Scholar] [CrossRef]
- Vallespín, E.; Palomares, B.M.; Mori, M.Á.; Martín, R.; García-Miñaúr, S.; Fernández, L.; de Torres, M.L.; García-Santiago, F.; Mansilla, E.; Santos, F.; et al. Customized high-resolution CGH-array for clinical diagnosis reveals additional genomic imbalances in previous well-defined pathological samples. Am. J. Med. Genet. A 2013, 161, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Talamantes, A.K.; Mori, M.Á.; Santos-Simarro, F.; García-Miñaur, S.; Mansilla, E.; Tenorio, J.A.; Peña, C.; Adan, C.; Fernández-Elvira, M.; Rueda, I.; et al. Chromosomal Microarray in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes 2023, 29, 820. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Torrico, B.; Hervás, A.; Valdés-Mas, R.; Tristán-Noguero, A.; Padillo, V.; Maristany, M.; Salgado, M.; Arenas, C.; Puente, X.S.; et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 2014, 19, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Arteche-López, A.; Gómez Rodríguez, M.J.; Sánchez Calvin, M.T.; Quesada-Espinosa, J.F.; Lezana Rosales, J.M.; Palma Milla, C.; Gómez-Manjón, I.; Hidalgo Mayoral, I.; de la Fuente, R.P.; de Bustamante, A.D.; et al. Towards a Change in the Diagnostic Algorithm of Autism Spectrum Disorders: Evidence Supporting Whole Exome Sequencing as a First-Tier Test. Genes 2021, 12, 560. [Google Scholar] [CrossRef]
- Martinez-Granero, F.; Blanco-Kelly, F.; Sanchez-Jimeno, C.; Avila-Fernandez, A.; Arteche, A.; Bustamante-Aragones, A.; Rodilla, C.; Rodríguez-Pinilla, E.; Riveiro-Alvarez, R.; Tahsin-Swafiri, S.; et al. Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. NPJ Genom. Med. 2021, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Codina-Solà, M.; Rodríguez-Santiago, B.; Homs, A.; Santoyo, J.; Rigau, M.; Aznar-Laín, G.; Del Campo, M.; Gener, B.; Gabau, E.; Botella, M.P.; et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol. Autism 2015, 6, 6–21. [Google Scholar] [CrossRef]
- Lossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, É.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015, 23, 1505–1512. [Google Scholar] [CrossRef]
- Xiong, J.; Chen, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Neurological Diseases with Autism Spectrum Disorder: Role of ASD Risk Genes. Front. Neurosci. 2019, 11, 349. [Google Scholar] [CrossRef]
- Li, J.; You, Y.; Yue, W.; Jia, M.; Yu, H.; Lu, T.; Wu, Z.; Ruan, Y.; Wang, L.; Zhang, D. Genetic Evidence for Possible Involvement of the Calcium Channel Gene CACNA1A in Autism Pathogenesis in Chinese Han Population. PLoS ONE 2015, 13, e0142887. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016, 13, e347ra94. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, J.; Okamoto, N.; Tohyama, J.; Kato, M.; Arai, H.; Funahashi, O.; Tsurusaki, Y.; Nakashima, M.; Kawashima, H.; Saitsu, H.; et al. De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy. Clin. Genet. 2015, 87, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Wang, H.; Song, Z.; Yin, X.; Wang, Y. EEF1A2 mutations in epileptic encephalopathy/intellectual disability: Understanding the potential mechanism of phenotypic variation. Epilepsy Behav. 2020, 105, 106955. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Adegbola, A.; Musante, L.; Callewaert, B.; Maciel, P.; Hu, H.; Isidor, B.; Picker-Minh, S.; Le Caignec, C.; Delle, C.B.; Vanakker, O.; et al. Redefining the MED13L syndrome. Eur. J. Hum. Genet. 2015, 23, 1308–1317. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Bookman, M.L.; Howe, J.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Reissner, C.; Klose, M.; Fairless, R.; Missler, M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc. Natl. Acad. Sci. USA 2008, 30, 15124–15129. [Google Scholar] [CrossRef] [PubMed]
- Barnby, G.; Abbott, A.; Sykes, N.; Morris, A.; Weeks, D.E.; Mott, R.; Lamb, J.; Bailey, A.J.; Monaco, A.P.; International Molecular Genetics Study of Autism Consortium. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: Evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 2005, 76, 950–966. [Google Scholar] [CrossRef]
- González, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Parra, A.; Rabin, R.; Pappas, J.; Pascual, P.; Cazalla, M.; Arias, P.; Gallego-Zazo, N.; Santana, A.; Arroyo, I.; Artigas, M.; et al. Clinical Heterogeneity and Different Phenotypes in Patients with SETD2 Variants: 18 New Patients and Review of the Literature. Genes 2023, 14, 1179. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wang, Y.; Li, C.; Mei, L.; Zhou, B.; Li, D.; Li, H.; Xu, Q.; Xu, X. Targeted sequencing and clinical strategies in children with autism spectrum disorder: A cohort study. Front. Genet. 2023, 14, 1083779. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; He, L.; Li, H.; Ding, Y.; Zhang, K.; Li, D.; Zhu, G.; Wu, B.; Xu, X.; Xu, Q. Clinical Targeted Panel Sequencing Analysis in Clinical Evaluation of Children with Autism Spectrum Disorder in China. Genes 2022, 13, 1010. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, X.; Liu, X.; Shen, L.; Wang, K.; Fan, Y.; Sun, Y.; Luo, X.; Liu, H.; Wang, L.; et al. Genetic Diagnostic Evaluation of Trio-Based Whole Exome Sequencing Among Children with Diagnosed or Suspected Autism Spectrum Disorder. Front. Genet. 2018, 9, 594. [Google Scholar] [CrossRef]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Mol. Autism 2019, 10, 33. [Google Scholar] [CrossRef]
- Kalsner, L.; Twachtman-Bassett, J.; Tokarski, K.; Stanley, C.; Dumont-Mathieu, T.; Cotney, J.; Chamberlain, S. Genetic testing including targeted gene panel in a diverse clinical population of children with autism spectrum disorder: Findings and implications. Mol. Genet. Genom. Med. 2018, 6, 171–185. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Correction: Meta-Analysis and Multidisciplinary Consensus Statement: Exome Sequencing Is a First-Tier Clinical Diagnostic Test for Individuals with Neurodevelopmental Disorders. Genet. Med. 2020, 22, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, P.; Zhou, X.; Astrovskaya, I.; Turner, T.N.; Wang, T.; Brueggeman, L.; Barnard, R.; Hsieh, A.; Snyder, L.G.; Muzny, D.M.; et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med. 2019, 4, 19. [Google Scholar] [CrossRef]
- Ni Ghralaigh, F.; Gallagher, L.; and Lopez, L.M. Autism spectrum disorder genomics: The progress and potential of genomic technologies. Genomics 2020, 112, 5136–5142. [Google Scholar] [CrossRef]
- Stefanski, A.; Calle-López, Y.; Leu, C.; Pérez-Palma, E.; Pestana-Knight, E.; Lal, D. Clinical Sequencing Yield in Epilepsy, Autism Spectrum Disorder, and Intellectual Disability: A Systematic Review and Meta-Analysis. Epilepsia 2021, 62, 143–151. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
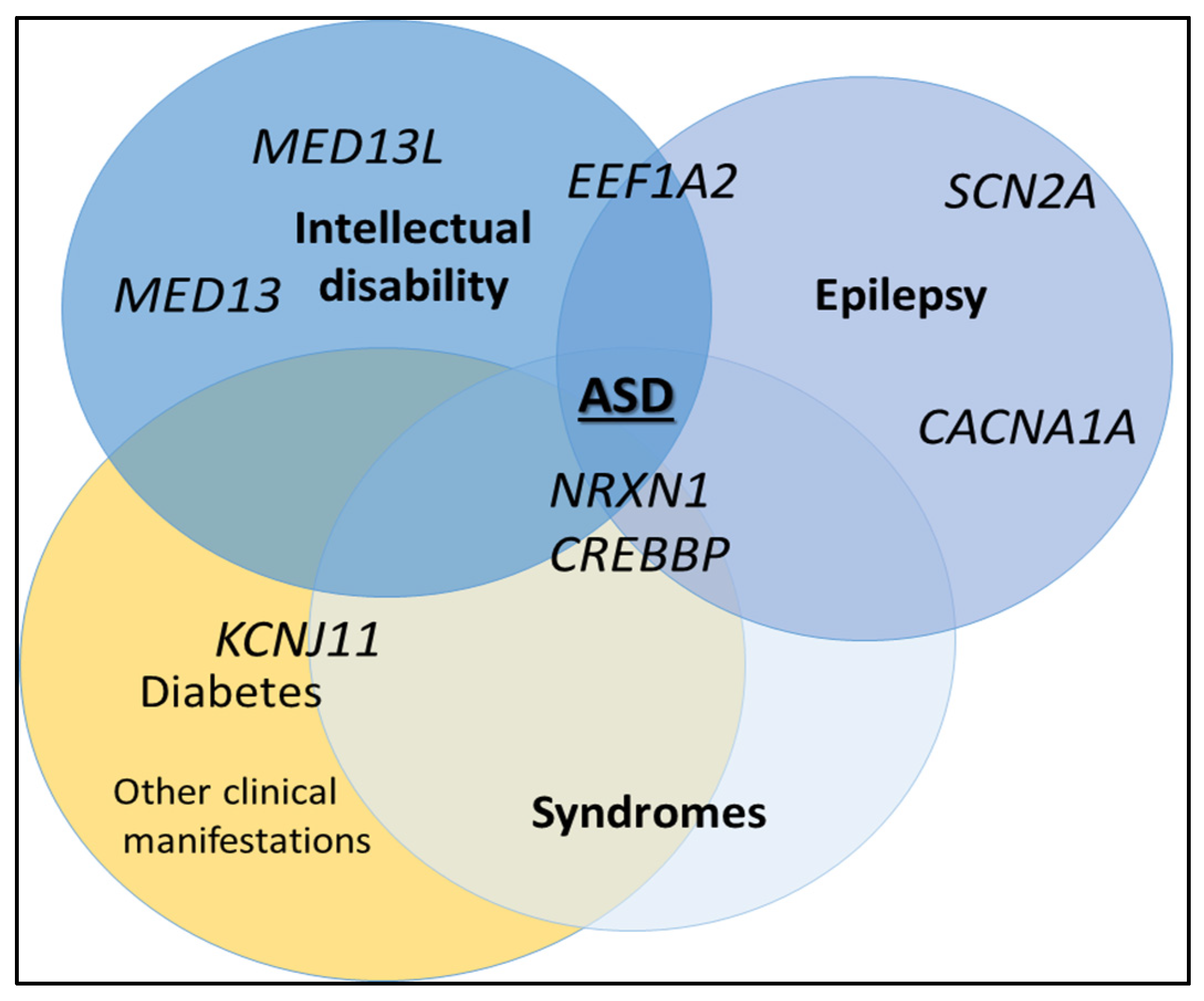{kind=link}
{kind=link}
| Patient/ Sex | Clinical Features in the Patient | Gen/Exon | cHGVS/ pHGVS/ Zygosity | Protein Effect dBSNP | OMIM (Gene Mainly Associated with) | Frequencies (gnomAD) | ACMG Classification | Conservation Predictors |
|---|---|---|---|---|---|---|---|---|
| AUT23 male | ASD | MED13L 5/31 | NM_015335.5: c.572del (p.Leu191Ter) heterozygous | frameshift | # 616789 Impaired intellectual development and distinctive facial features with or without cardiac defects (AD) | Not Found | LP; PVS1, PM2 (9 pts; 9P-0B) | PhastCons100 Way: 1.00 PyloP100way: 5.488; 5.344 GERP RS: 4.72 median |
| AUT99 male | ASD | KCNJ11 1/1 NBEAL1 13/55 | NM_0000525.4: c.325C>A p.(Pro109Thr) heterozygous NM_001114132.2: c.1741del p.Val581CfsTer20 heterozygous | missense rs758228551 frameshift | # 610582 Diabetes mellitus, transient neonatal 3 (AD) # 618856 Diabetes, permanent neonatal 2, with or without neurologic features (AD) # 601820 Hyperinsulinemic hypoglycemia, familial, 2 (AD y AR) # 616329 Maturity-onset diabetes of the young, type 13 (AD) none | Exomes: f = 0.00000398; not in Eur. NF Genomes: Not Found Not Found | VUS-LP; PM2, PP2, PP3 (3 pts; 3P-0B) PP3 pathogenic supporting: CADD, MutPred, MVP, PROVEAN, SIFT4G, DANN, MetaRNN, BayesDeladdAF VUS-LP; PVS1, PM2 (5 pts; 5P-0B) | PhastCons100 Way: 1.00 PyloP100way: 5.366 GERP RS: 3.99 PhastCons100 Way: 1.00 PyloP100way: 6.062 GERP RS: 5.519 |
| AUT108 male | ASD | SCN2A 28/28 | NM_001040142.2: c.5890G>A p.Asp1964Asn heterozygous | missense | # 613721 Developmental and epileptic encephalopathy 11 (AD) # 618924 Episodic ataxia, type 9 (AD) # 607745 Seizures, benign familial infantile, 3 (AD) | Exomes: f = 0.00000398; not in Eur. NF Genomes: Not Found | VUS-LP; PM2, PP2, PP3 (3 pts; 3P-0B) PP3 pathogenic supporting: CADD, EIGEN PC, MVP, PROVEAN, FATHAMM-MKL, DANN, MetaLR, MetaSVM | PhastCons100 Way: 1.00 PyloP100way: 8.017 GERP RS: 5.73 |
| AUT114 male | ASD | CHD7 16/38 | NM_017780.4: c.3973T>C p.Yyr1325His heterozygous | Missense rs377535841 | # 214800 CHARGE syndrome (AD) # 612370 Hypogonadotropic hypogonadism 5 with or without anosmia (AD) | Exomes: f = 0.0000684 Genomes: f = 0.0000637 | VUS-LP; PM1,PM5, PP3, BS2 (4 pts; 5P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, LRT, LIST-S2, M_CAP, PROVEAN, SIFT DANN, MetaRNN, REVEL, BayesDel no AF, BayesDel addAF CliVar (conflicting VUS:5, LB:3); UNIPROT (LP); Varsome (VUS) | PhastCons100 Way: 1.00 PyloP100way: 8.042 GERP RS: 5.8 |
| AUT115 male | ASD | CHD7 16/38 | NM_017780.4: c.3973T>C p.Yyr1325His heterozygous | Missense rs377535841 | # 214800 CHARGE syndrome (AD) # 612370 Hypogonadotropic hypogonadism 5 with or without anosmia (AD) | Exomes: f = 0.0000684 Genomes: f = 0.0000637 | VUS-LP; PM1,PM5, PP3, BS2 (4 pts; 5P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, LRT, LIST-S2, M_CAP, PROVEAN, SIFT DANN, MetaRNN, REVEL, BayesDel no AF, BayesDel addAF CliVar (conflicting VUS:5, LB:3); UNIPROT (LP); Varsome (VUS) | PhastCons100 Way: 1.00 PyloP100way: 8.042 GERP RS: 5.8 |
| AUT117 male | ASD | CACNA1A 31/48 | NM_001127221.2: c.4880G>A p.Arg1627His heterozygous | missense rs777769751 | # 617106 Developmental and epileptic encephalopathy 42 (AD) # 108500 Episodic ataxia, type 2 (AD) # 141500 Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia (AD) # 183086 Spinocerebellar ataxia 6 (AD) | Exomes: f = 0.0000443; in Eur. NF Genomes: Not Found | VUS-LP; PM2, PP3 (3 pts; 3P-0B) PP3 pathogenic supporting: CADD, M-CAP, PrimateAI MVP, PROVEAN, FATHAMM, DANN, MetaLR, MetaRNN, MetaSVN, REVEL; ClinVAR (VUS) | PhastCons100 Way: 1.00 PyloP100way: 4.918 GERP RS: 3.63 |
| AUT142 male | ASD | CACNA1D 42/49 | NM_001128840.3: c.4967G>A p.Arg1656His heterozygous | Missense rs890934509 | # 615474 Primary aldosteronism, seizures, and neurologic abnormalities (AD) # 614896 Sinoatrial node dysfunction and deafness (AR) | Exomes: f = 0.00000398; in Eur. NF Genomes: not found | VUS-LP; PM2, PP3 (3 pts; 3P-0B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, PrimateAI, FATHAMM, FATHAMM-XF, M-CAP, LIST-S2, PROVEAN, FATHAMM-MKL, DANN, MetaLR, MetaRNN, MetaSVN, REVEL, BAyesDel no AF, BayesDel addAF | PhastCons100 Way: 0.996 PyloP100way: 2.496 GERP RS: 2.97 |
| AUT149 male | Asperger depression | EEF1A2 4/8 | NM_001958.5: c.479C>T p.Pro160Leu heterozygous | missense | # 616409 Developmental and epileptic encephalopathy 33 (AD) # 616393 Intellectual developmental disorder, autosomal dominant 38 (AD) | Not Found | VUS-LP; PM2, PP2, PP3 (3 pts; 3P-0B) PP3 pathogenic supporting: CADD, PrimateAI, LRT, PROVEAN, SIFT, FATHAMM-MKL, DANN, MetaRNN | PhastCons100 Way: 1.00 PyloP100way: 9.659 GERP RS: 3.866 |
| AUT157 male | ASD | CREBBP 17/30 | NM_004380.3: c.3559C>T p.Arg1187Ter heterozygous | nonsense | # 618332 Menke–Hennekam syndrome 1 (AD) # 180849 Rubinstein–Taybi syndrome 1 (AD) | Not Found | LP; PVS1, PM2 (9 pts; 9P-0B) | PhastCons100 Way: 1.00 PyloP100way: 7.389 GERP RS: 5.59 |
| AUT 187 male | ASD | SCN2A 16/27 | NM_001040142.2: c.2789A>C p.His930Pro heterozygous | missense | # 613721 Developmental and epileptic encephalopathy 11 (AD) # 618924 Episodic ataxia, type 9 (AD) # 607745 Seizures, benign familial infantile, 3 (AD) | Not Found | LP; PM1, PM2, PP3 (7 pts; 3P-0B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, DEOGEN2, MVP, PROVEAN, MVP, M-CAP, MutPred, FATHAMM, Mutation Assesor, PrimateAI, SIFT, SIFT4G FATHAMM-MKL, FATHAMM-XF, DANN, MetaLR, MetaSVM, REVEL, MetaRNN, BayesDel no AF, BayesDel Addai | PhastCons100 Way: 1.00 PyloP100way: 9.198 GERP RS: 5.42 |
| AUT195 female | ASD ID Tall stature Hemi-hypertrophy | SETD2 15/21 CHD2 16/39 | NM_014159.7: c.6299A>G p.Asp2100Gly heterozygous NM_001271.4: c.1994C>T p.Pro665Leu heterozygous | Missense Missense | # 616831 Luscan–Lumish syndrome (AD) # 615369 Developmental and epileptic encephalopathy 94 (AD) | Exomes: f = 0.00000401, in Eur. NF Genomes: not found Exomes: f = 0.0000559, in Eur. NF Genomes: not found | VUS; PM2, BP1 (1 pts; 2P-1B) PP3 pathogenic supporting: CADD, DANN, FATHAMM-MKL LP; PM1, PM2, PP3, (7pts; 6P-0B) PP3 pathogenic supporting: CADD, DANN, DEOGEN2, EIGEN, EIGEN PC, MVP, PROVEAN, FATHAMM_MKL, M-CAP, PrimateAI, SIFT, SIFT4G, METARNN, BayesDel addAF, BayesDel no AF, MetaLR, MetaSVM, REVEL, ClinVar (conflicting) | PhastCons100 Way: 1.00 PyloP100way: 6.81 GERP RS: 4.84 PhastCons100 Way: 1.00 PyloP100way: 7.817 GERP RS: 5.82 |
| Patient/ Sex | Clinical Features in the Patient | Gen/Exon | cHGVS/ pHGVS/ Zygosity | Protein Effect dBSNP | OMIM (Gene Associated with.) | Frequencies (gnomAD) | ACMG Classification | Conservation Predictors |
|---|---|---|---|---|---|---|---|---|
| AUT118 male | Asperger | MED13 2/30 | NM_005121.3: c.124C>T (p.Pro42Ser)/ heterozygous | missense rs778909357 | # 618009 Intellectual developmental disorder, autosomal dominant 61 (AD) | Exomes: f = 0.0000579; not in Eur.NF Genomes: Not Found | VUS; PM2, PP3 (2 pts; 2P-0B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, LRT, PrimateAI, MVP, PROVEAN, FATHAMM-XF, DANN, MetaRNN | PhastCons100 Way: 1.00 PyloP100way: 9.873 GERP RS: 5.67 |
| AUT120 male | ASD | NRXN1 1/7 | NM_004801.5: c.77_79dup p.Gly26dup heterozygous | In-frame Insertion rs766368745 | #614325 Pitt–Hopkins-like syndrome 2 (AR) | Not Found | VUS; PM4, PM2 (3 pts; 3P-0B) No info | PhastCons100 Way: 1.00 PyloP100way: 4.046, 5.708, 1, 43, 3.125 GERP RS: 5.05 median |
| AUT140 male | ASD ADHD epilepsy | CEP290 19/54 | c.1834C>T p.Leu612Phe heterozygous | Missense | 615991 Bardet–Biedl syndrome 14 (AR) # 610188 Joubert syndrome 5 (AR) # 611755 Leber congenital amaurosis 10 # 611134 Meckel syndrome 4 (AR) # 610189 Senior–Loken syndrome 6 (AR) | Not Found | VUS; PM2, PP3 (2 pts; 2P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, FATHAMM-MKL, DANN | PhastCons100 Way: 1.00 PyloP100way: 7.562 GERP RS: 5.349 |
| AUT147 male | ASD | TCF20 2/6 | NM:_001378418.1: c.454T>G p.Tyr152Asp heterozygous | Missense | # 618430 Developmental delay with variable intellectual impairment and behavioral abnormalities (AD) | Not Found | VUS; PM2, PP3 (1 pts; 2P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, PrimateAI, SIFT, SIFT4G, FATHAMM-MKL, DANN, BAyesDel no AF, BayesDel addAF | PhastCons100 Way: 1.00 PyloP100way: 8.785 GERP RS: 5.519 |
| AUT152 male | ASD PMD | SPTBN1 25/36 ZEB2 7/9 | NM_003128.3: c.5014C>T p.Arg1672Trp heterozygous NM_014795.4: c.1769T>C p.Leu590Pro heterozygous | Missense rs755243358 Missense | # 619475 Developmental delay, impaired speech, and behavioral abnormalities (AD) # 235730 Mowat–Wilson syndrome (AD) | Exomes: f = 0.0000161 Genomes: f = 0.0000319 Not Found | VUS; PM2, PP3 (2 pts; 2P-0B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, PrimateAI, SIFT, SIFT4G, FATHAMM-MKL, DANN, BAyesDel no AF, BayesDel addAF VUS; PM2, PP3, BP1 (1 pts; 2P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, PrimateAI, LRT, MCAP, MutPred, LIST-S2 FATHAMM-MKL, DANN, BAyesDel no AF, BayesDel addAF REVEL, MetaRNN | PhastCons100 Way: 1.00 PyloP100way: 8.785 GERP RS: 5.519 PhastCons100 Way: 1.00 PyloP100way: 9.339 GERP RS: 5.75 |
| AUT161 male | ASD ID | CACNA1A 46/48 | NM_023035.3: c.6512G>A p.Arg2171His heterozygous | missense rs727503832 | # 617106 Developmental and epileptic encephalopathy 42 (AD) # 108500 Episodic ataxia, type 2 (AD) # 141500 Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia (AD) # 183086 Spinocerebellar ataxia 6 (AD) | Exomes: f = 0.000326; in Eur.NF Genomes: f = 0.000195; in Eur.NF | VUS; PM2, PP3 (2 pts; 2P-0B) PP3 pathogenic supporting: CADD, LIST-S2, PrimateAI, M-CAP, FATHAMM, DANN, MetaLR, BayesDel addAF ClinVAR (conflicting, 1star) | PhastCons100 Way: 1.00 PyloP100way: 5.001 GERP RS: 3.38 |
| AUT171 male | ASD | KMT2D | NM_003482.4: c.13885A>C p.Thr4629Pro heterozygous | Missense | # 147920 Kabuki Syndrome 1 | Not Found | VUS; PM2, PP3, BP1 (1 pts; 2P-1B) PP3 pathogenic supporting: CADD, EIGEN, EIGEN PC, PrimateAI, FATHAMM-XF, MCAP, MutPred, PROVEAN FATHAMM-MKL, DANN, AF, BayesDel addAF; LOVD(VUS) | PhastCons100 Way: 1.00 PyloP100way: 7.972 GERP RS: 5.579 |
| AUT183 male | Asperger | CHD1 3/35 | (NM_001270.4): c.315G>C p.Gln105His heterozygous | Missense rs906013276 | # 617682 Pilarowski–Bjornsson syndrome (AD) | Not Found | VUS; PM2, PP2, (2 pts; 2P-0B) PP3 pathogenic supporting: CADD, DANN, many others uncertain | PhastCons100 Way:1.00 PyloP100way: 1.34 GERP RS: 4.789 |
| AUT 221 male | ASD | CACNA1A 1/48 | NM_023035.3: c.115G>A p.G39S p.Gly39Ser heterozygous | missense | # 617106 Developmental and epileptic encephalopathy 42 (AD) # 108500 Episodic ataxia, type 2 (AD) # 141500 Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia (AD) # 183086 Spinocerebellar ataxia 6 (AD) | Not Found | VUS; PM2, PP3 (2 pts; 2P-0B) PP3 pathogenic supporting: CADD, PrimateAI, M-CAP, FATHAMM, DANN, MetaLR | PhastCons100 Way: 0.996 PyloP100way: 2.496 GERP RS: 2.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval-Talamantes, A.K.; Tenorio-Castaño, J.A.; Santos-Simarro, F.; Adán, C.; Fernández-Elvira, M.; García-Fernández, L.; Muñoz, Y.; Lapunzina, P.; Nevado, J. NGS Custom Panel Implementation in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes 2023, 14, 2091. https://doi.org/10.3390/genes14112091
Sandoval-Talamantes AK, Tenorio-Castaño JA, Santos-Simarro F, Adán C, Fernández-Elvira M, García-Fernández L, Muñoz Y, Lapunzina P, Nevado J. NGS Custom Panel Implementation in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes. 2023; 14(11):2091. https://doi.org/10.3390/genes14112091
Chicago/Turabian StyleSandoval-Talamantes, Ana Karen, Jair Antonio Tenorio-Castaño, Fernando Santos-Simarro, Carmen Adán, María Fernández-Elvira, Laura García-Fernández, Yolanda Muñoz, Pablo Lapunzina, and Julián Nevado. 2023. "NGS Custom Panel Implementation in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital" Genes 14, no. 11: 2091. https://doi.org/10.3390/genes14112091
APA StyleSandoval-Talamantes, A. K., Tenorio-Castaño, J. A., Santos-Simarro, F., Adán, C., Fernández-Elvira, M., García-Fernández, L., Muñoz, Y., Lapunzina, P., & Nevado, J. (2023). NGS Custom Panel Implementation in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes, 14(11), 2091. https://doi.org/10.3390/genes14112091