
Double Heterozygous Pathogenic Variants in the LOX and PKD1 Genes in a 5-Year-Old Patient with Thoracic Aortic Aneurysm and Polycystic Kidney Disease

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Faggion Vinholo, T.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2019 Update and Clinical Implications. Aorta 2019, 7, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, A.J.; Kostiuk, V.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2018 Update and Clinical Implications. Aorta 2018, 6, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Pomianowski, P.; Elefteriades, J.A. The genetics and genomics of thoracic aortic disease. Ann. Cardiothorac. Surg. 2013, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- Poninska, J.K.; Bilinska, Z.T.; Franaszczyk, M.; Michalak, E.; Rydzanicz, M.; Szpakowski, E.; Pollak, A.; Milanowska, B.; Truszkowska, G.; Chmielewski, P.; et al. Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: Diagnostic yield, novel mutations and genotype phenotype correlations. J. Transl. Med. 2016, 14, 115. [Google Scholar] [CrossRef]
- Takeda, N.; Komuro, I. Genetic basis of hereditary thoracic aortic aneurysms and dissections. J. Cardiol. 2019, 74, 136–143. [Google Scholar] [CrossRef]
- Salazar-Mendiguchía, J.; Ochoa, J.P.; Palomino-Doza, J.; Domínguez, F.; Díez-López, C.; Akhtar, M.; Ramiro-León, S.; Clemente, M.M.; Pérez-Cejas, A.; Robledo, M.; et al. Mutations in TRIM63 cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart 2020, 106, 1342–1348. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- de Vries, B.B.; Pals, G.; Odink, R.; Hamel, B.C. Homozygosity for a FBN1 missense mutation: Clinical and molecular evidence for recessive Marfan syndrome. Eur. J. Hum. Genet. 2007, 15, 930–935. [Google Scholar] [CrossRef]
- Hilhorst-Hofstee, Y.; Rijlaarsdam, M.E.; Scholte, A.J.; Swart-van den Berg, M.; Versteegh, M.I.; van der Schoot-van Velzen, I.; Schäbitz, H.J.; Bijlsma, E.K.; Baars, M.J.; Kerstjens-Frederikse, W.S.; et al. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum. Mutat. 2010, 31, E1915–E1927. [Google Scholar] [CrossRef]
- Iglesias, C.G.; Torres, V.E.; Offord, K.P.; Holley, K.E.; Beard, C.M.; Kurland, L.T. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am. J. Kidney Dis. 1983, 2, 630–639. [Google Scholar] [CrossRef]
- Ecder, T.; Schrier, R.W. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat. Rev. Nephrol. 2009, 5, 221–228. [Google Scholar] [CrossRef]
- Chapman, A.B.; Rubinstein, D.; Hughes, R.; Stears, J.C.; Earnest, M.P.; Johnson, A.M.; Gabow, P.A.; Kaehny, W.D. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 1992, 327, 916–920. [Google Scholar] [CrossRef]
- Ruggieri, P.M.; Poulos, N.; Masaryk, T.J.; Ross, J.S.; Obuchowski, N.A.; Awad, I.A.; Braun, W.E.; Nally, J.; Lewin, J.S.; Modic, M.T. Occult intracranial aneurysms in polycystic kidney disease: Screening with MR angiography. Radiology 1994, 191, 33–39. [Google Scholar] [CrossRef]
- Nunes, R.; Gouveia, E.M.R.; Almeida, A.G.; de Almeida, E.; Pinto, F.J.; Pedro, L.M.; Caldeira, D. Does autosomal dominant polycystic kidney disease increase the risk of aortic aneurysm or dissection: A point of view based on a systematic review and meta-analysis. J. Nephrol. 2022, 35, 1585–1593. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Preventza, O.; Hamilton Black, J., 3rd; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 80, e223–e393. [Google Scholar] [CrossRef]
- Śmigiel, R.; Biela, M.; Szmyd, K.; Błoch, M.; Szmida, E.; Skiba, P.; Walczak, A.; Gasperowicz, P.; Kosińska, J.; Rydzanicz, M.; et al. Rapid Whole-Exome Sequencing as a Diagnostic Tool in a Neonatal/Pediatric Intensive Care Unit. J. Clin. Med. 2020, 9, 2220. [Google Scholar] [CrossRef]
- Pettersen, M.D.; Du, W.; Skeens, M.E.; Humes, R.A. Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: An echocardiographic study. J. Am. Soc. Echocardiogr. 2008, 21, 922–934. [Google Scholar] [CrossRef]
- Daubeney, P.E.; Blackstone, E.H.; Weintraub, R.G.; Slavik, Z.; Scanlon, J.; Webber, S.A. Relationship of the dimension of cardiac structures to body size: An echocardiographic study in normal infants and children. Cardiol. Young 1999, 9, 402–410. [Google Scholar] [CrossRef]
- Devereux, R.B.; de Simone, G.; Arnett, D.K.; Best, L.G.; Boerwinkle, E.; Howard, B.V.; Kitzman, D.; Lee, E.T.; Mosley, T.H., Jr.; Weder, A.; et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥15 years of age. Am. J. Cardiol. 2012, 110, 1189–1194. [Google Scholar] [CrossRef]
- Rozendaal, L.; Groenink, M.; Naeff, M.S.; Hennekam, R.C.; Hart, A.A.; van der Wall, E.E.; Mulder, B.J. Marfan syndrome in children and adolescents: An adjusted nomogram for screening aortic root dilatation. Heart 1998, 79, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.A.; Li, Y.; Rizzo, J.A.; Charilaou, P.; Saeyeldin, A.; Velasquez, C.A.; Mansour, A.M.; Bin Mahmood, S.U.; Ma, W.G.; Brownstein, A.J.; et al. Height alone, rather than body surface area, suffices for risk estimation in ascending aortic aneurysm. J. Thorac. Cardiovasc. Surg. 2018, 155, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ. Res. 2016, 118, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Van Gucht, I.; Krebsova, A.; Diness, B.R.; Laga, S.; Adlam, D.; Kempers, M.; Samani, N.J.; Webb, T.R.; Baranowska, A.A.; Van Den Heuvel, L.; et al. Novel LOX Variants in Five Families with Aortic/Arterial Aneurysm and Dissection with Variable Connective Tissue Findings. Int. J. Mol. Sci. 2021, 22, 7111. [Google Scholar] [CrossRef]
- Atsawasuwan, P.; Mochida, Y.; Katafuchi, M.; Kaku, M.; Fong, K.S.; Csiszar, K.; Yamauchi, M. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J. Biol. Chem. 2008, 283, 34229–34240. [Google Scholar] [CrossRef]
- Neumann, H.P.; Jilg, C.; Bacher, J.; Nabulsi, Z.; Malinoc, A.; Hummel, B.; Hoffmann, M.M.; Ortiz-Bruechle, N.; Glasker, S.; Pisarski, P.; et al. Epidemiology of autosomal-dominant polycystic kidney disease: An in-depth clinical study for south-western Germany. Nephrol. Dial. Transpl. 2013, 28, 1472–1487. [Google Scholar] [CrossRef]
- Thivierge, C.; Kurbegovic, A.; Couillard, M.; Guillaume, R.; Coté, O.; Trudel, M. Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell Biol. 2006, 26, 1538–1548. [Google Scholar] [CrossRef]
- Peczkowska, M.; Januszewicz, A.; Grzeszczak, W.; Moczulski, D.; Janaszek-Sitkowska, H.; Kabat, M.; Biederman, A.; Hendzel, P.; Prejbisz, A.; Cendrowska-Demkow, I.; et al. The coexistence of acute aortic dissection with autosomal dominant polycystic kidney disease--description of two hypertensive patients. Blood Press. 2004, 13, 283–286. [Google Scholar] [CrossRef]
- Sung, P.H.; Yang, Y.H.; Chiang, H.J.; Chiang, J.Y.; Chen, C.J.; Liu, C.T.; Yu, C.M.; Yip, H.K. Risk of aortic aneurysm and dissection in patients with autosomal-dominant polycystic kidney disease: A nationwide population-based cohort study. Oncotarget 2017, 8, 57594–57604. [Google Scholar] [CrossRef]
- Perrone, R.D.; Malek, A.M.; Watnick, T. Vascular complications in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 589–598. [Google Scholar] [CrossRef]
- Spinelli, L.; Giugliano, G.; Esposito, G. Cardiac Involvement in Autosomal Dominant Polycystic Kidney Disease. Cardiogenetics 2021, 11, 39–49. [Google Scholar] [CrossRef]
- Liu, D.; Wang, C.J.; Judge, D.P.; Halushka, M.K.; Ni, J.; Habashi, J.P.; Moslehi, J.; Bedja, D.; Gabrielson, K.L.; Xu, H.; et al. A Pkd1-Fbn1 genetic interaction implicates TGF-β signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kwartler, C.S.; Gong, L.; Chen, J.; Wang, S.; Kulmacz, R.; Duan, X.Y.; Janda, A.; Huang, J.; Kamm, K.E.; Stull, J.T.; et al. Variants of Unknown Significance in Genes Associated with Heritable Thoracic Aortic Disease Can Be Low Penetrant “Risk Variants”. Am. J. Hum. Genet. 2018, 103, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Tcheandjieu, C.; Xiao, K.; Tejeda, H.; Lynch, J.A.; Ruotsalainen, S.; Bellomo, T.; Palnati, M.; Judy, R.; Klarin, D.; Kember, R.L.; et al. High heritability of ascending aortic diameter and trans-ancestry prediction of thoracic aortic disease. Nat. Genet. 2022, 54, 772–782. [Google Scholar] [CrossRef]
- Disha, K.; Schulz, S.; Mierzwa, M.; Owais, T.; Girdauskas, E.; Kuntze, T. Double-Hit Mutations in Bicuspid Aortic Valve and Blunt Traumatic Acute Aortic Dissection. Ann. Thorac. Surg. 2021, 111, e5–e6. [Google Scholar] [CrossRef]
- Klarin, D.; Devineni, P.; Sendamarai, A.K.; Angueira, A.R.; Graham, S.E.; Shen, Y.H.; Levin, M.G.; Pirruccello, J.P.; Surakka, I.; Karnam, P.R.; et al. Genome-wide association study of thoracic aortic aneurysm and dissection in the Million Veteran Program. Nat. Genet. 2023, 55, 1106–1115. [Google Scholar] [CrossRef]
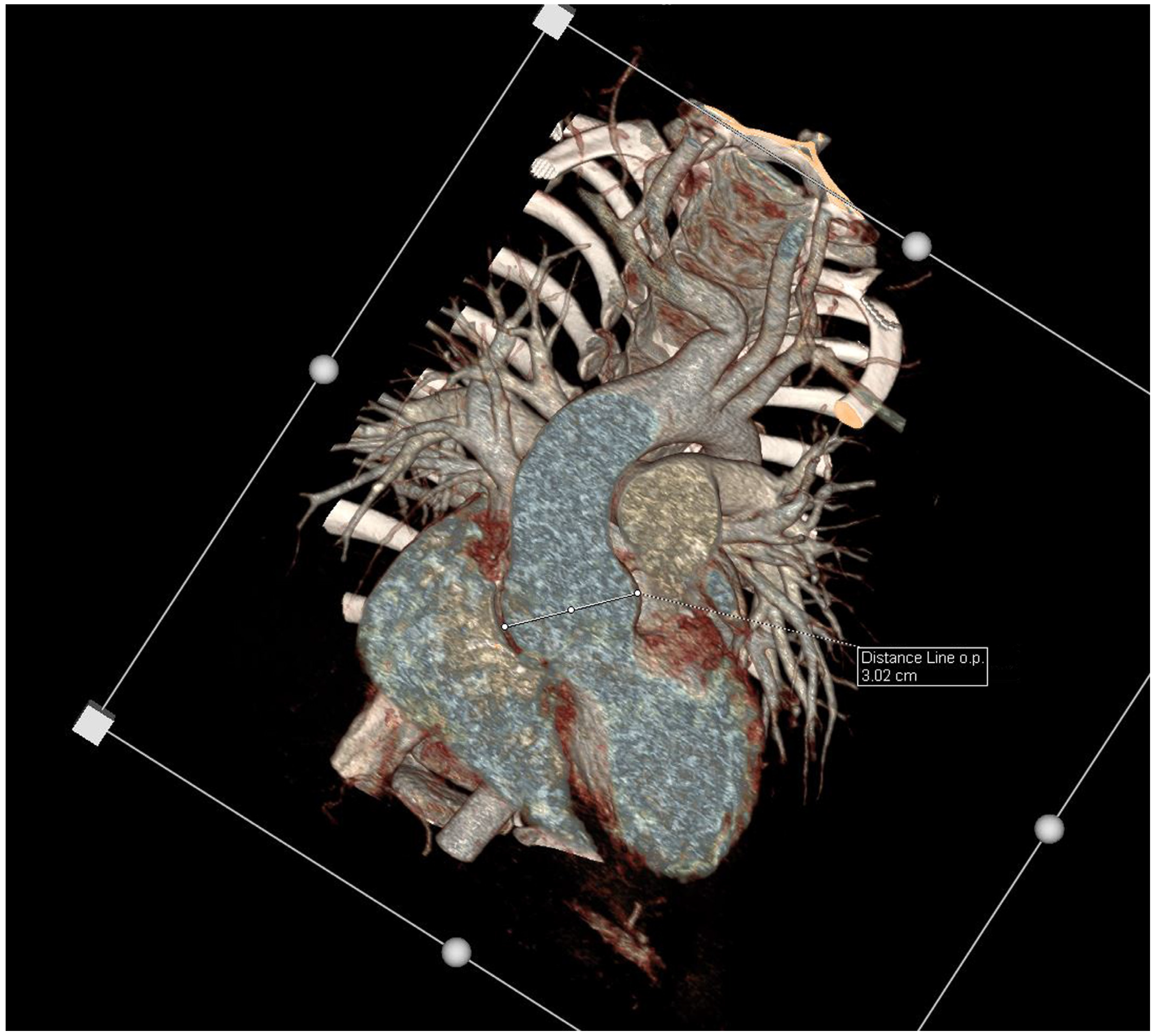

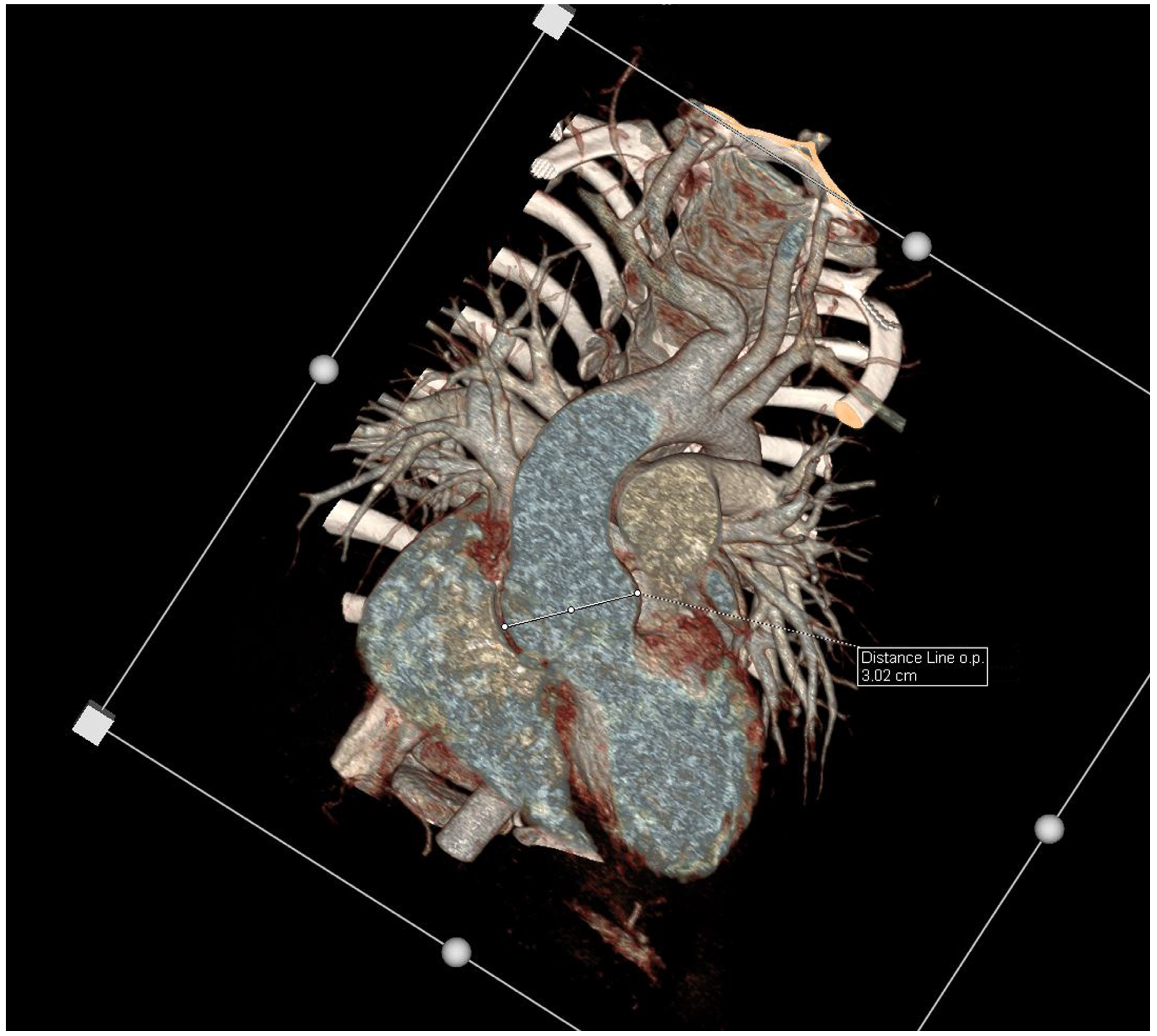{kind=link}
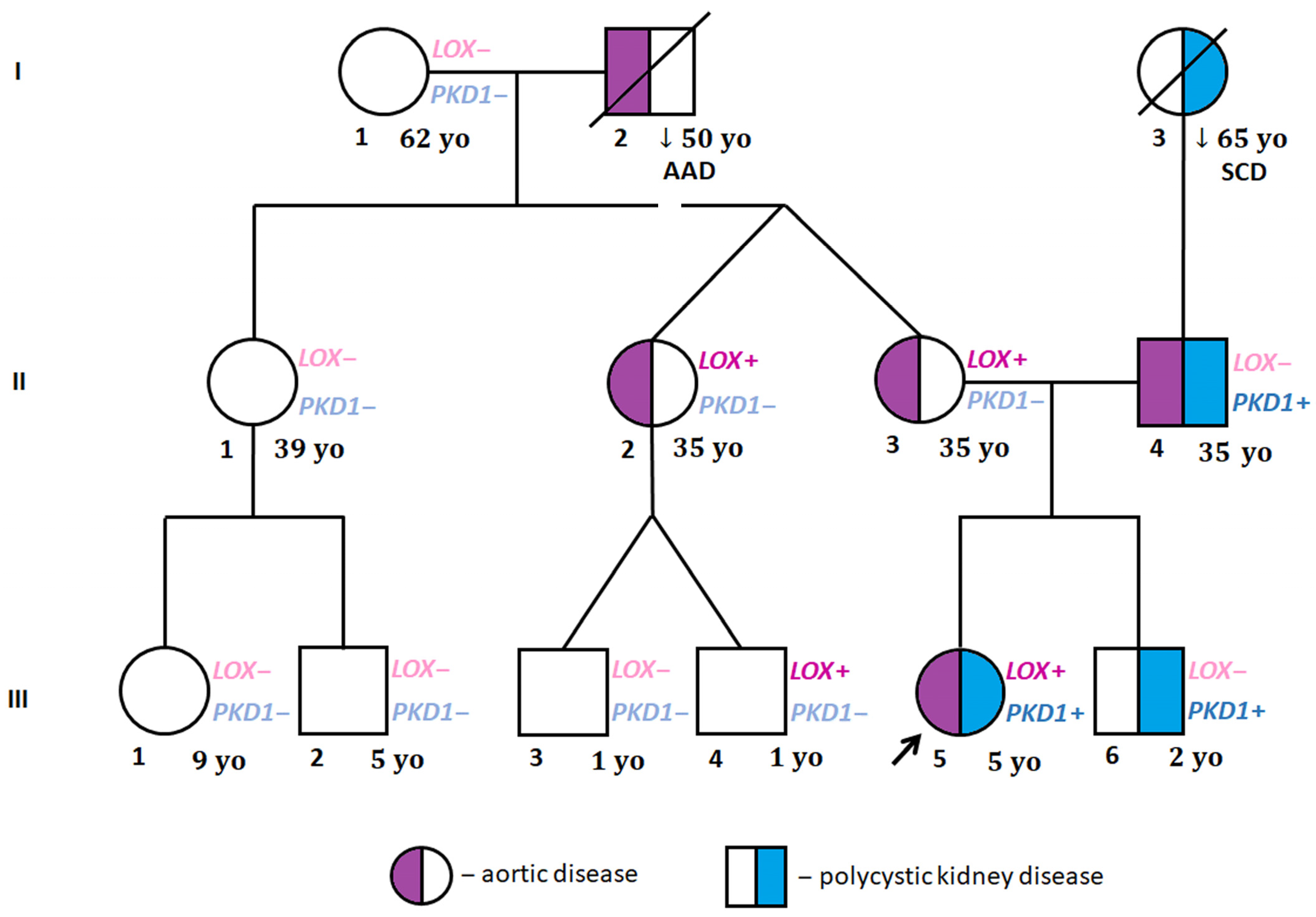
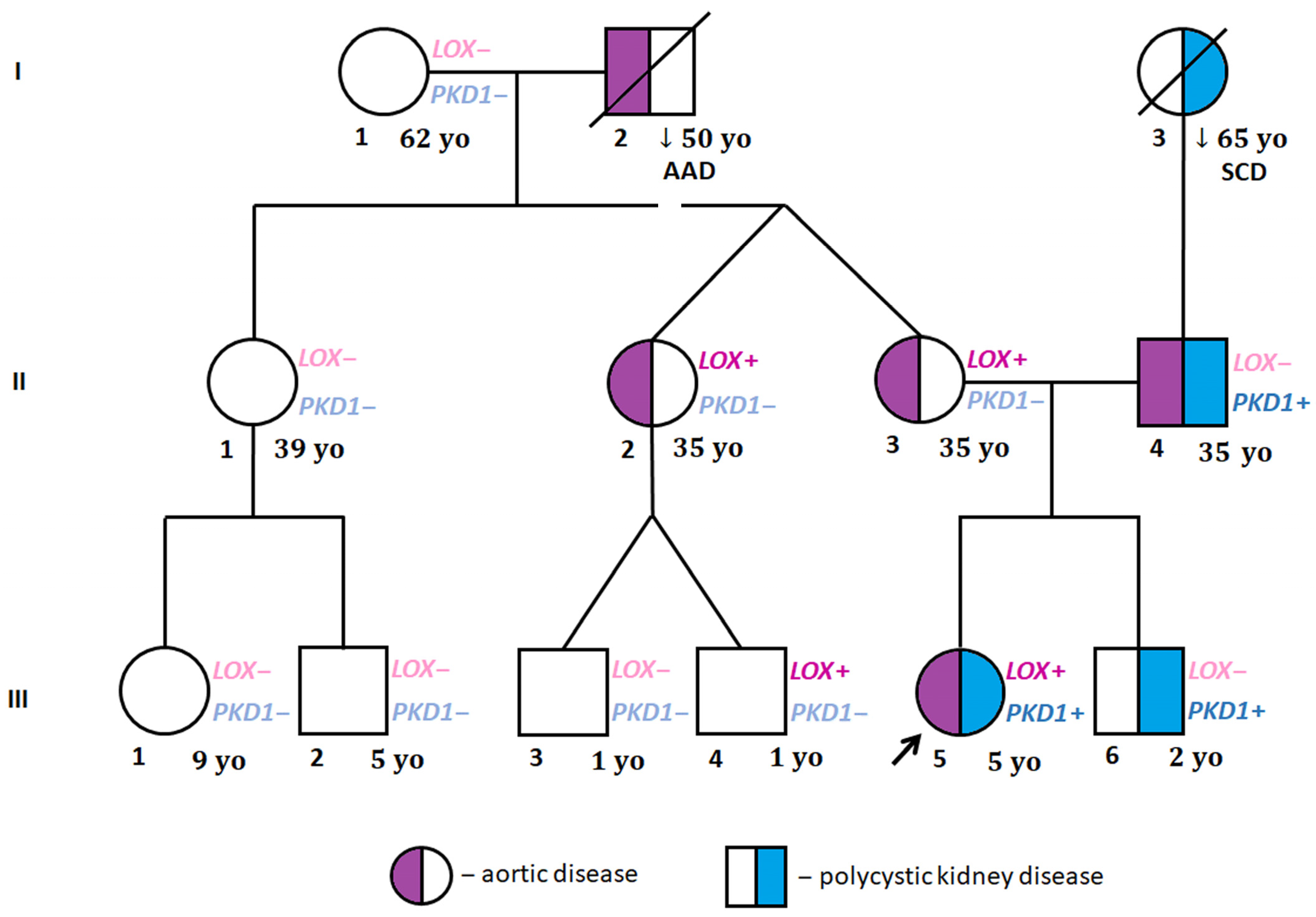{kind=link}
| Patient | Pedigree | Age at Examination (Years) | Weight (kg) | Height (m) | BSA | Aortic Root (mm) | Z-Score [20,21] | AHI [22] | Presence of Kidney Cysts |
|---|---|---|---|---|---|---|---|---|---|
| Proband | III 5 | 5.5 | 23 | 1.23 | 0.88 | 27 | 3.76 | 2.20 | Bilateral > 6 |
| Mother | II 3 | 35 | 73 | 1.84 | 1.95 | 39 | 3.05 | 2.12 | Excluded by CTA |
| Father | II 4 | 35 | 115 | 1.98 | 2.5 | 47 | 4.12 | 2.37 | Multiple single bilateral |
| Brother | III 6 | 2 | 9 | 0.74 | 0.44 | 13 | −0.47 | 1.75 | Bilateral > 10 |
| Paternal grandmother | I 3 | deceased at 65 (SCD) | N/A | N/A | N/A | N/A | N/A | N/A | History of kidney and liver cysts |
| Maternal aunt II | II 2 | 35 | 85 | 1.72 | 1.98 | 38 | 2.58 | 2.21 | Excluded by CTA |
| Cousin III | III 3 | 4 | 20 | 1.14 | 0.8 | 19 | −0.4 | 1.67 | Excluded by ultrasound |
| Cousin IV | III 4 | 4 | 20 | 1.11 | 0.78 | 19 | −0.3 | 1.71 | Excluded by ultrasound |
| Gene | Reference Sequence | Coding | Protein | Chromosome Position (hg38) | Type | Frequency in gnomAD | ClinVar | ACMG Classification (Varsome ver. 11.8.4) |
|---|---|---|---|---|---|---|---|---|
| LOX | NM_002317.7 | c.771T>G | p.Tyr257Ter | 5:122075511-A>C | Nonsense | 0 | Absent | Likely pathogenic |
| PKD1 | NM_000296.4 | c.8930C>T | p.Thr2977Ile | 16:2102832-G>A | Missense | 0 | Absent | Uncertain significance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponińska, J.K.; Pelczar-Płachta, W.; Pollak, A.; Jończyk-Potoczna, K.; Truszkowska, G.; Michałowska, I.; Szafran, E.; Bilińska, Z.T.; Bobkowski, W.; Płoski, R. Double Heterozygous Pathogenic Variants in the LOX and PKD1 Genes in a 5-Year-Old Patient with Thoracic Aortic Aneurysm and Polycystic Kidney Disease. Genes 2023, 14, 1983. https://doi.org/10.3390/genes14111983
Ponińska JK, Pelczar-Płachta W, Pollak A, Jończyk-Potoczna K, Truszkowska G, Michałowska I, Szafran E, Bilińska ZT, Bobkowski W, Płoski R. Double Heterozygous Pathogenic Variants in the LOX and PKD1 Genes in a 5-Year-Old Patient with Thoracic Aortic Aneurysm and Polycystic Kidney Disease. Genes. 2023; 14(11):1983. https://doi.org/10.3390/genes14111983
Chicago/Turabian StylePonińska, Joanna Kinga, Weronika Pelczar-Płachta, Agnieszka Pollak, Katarzyna Jończyk-Potoczna, Grażyna Truszkowska, Ilona Michałowska, Emilia Szafran, Zofia T. Bilińska, Waldemar Bobkowski, and Rafał Płoski. 2023. "Double Heterozygous Pathogenic Variants in the LOX and PKD1 Genes in a 5-Year-Old Patient with Thoracic Aortic Aneurysm and Polycystic Kidney Disease" Genes 14, no. 11: 1983. https://doi.org/10.3390/genes14111983
APA StylePonińska, J. K., Pelczar-Płachta, W., Pollak, A., Jończyk-Potoczna, K., Truszkowska, G., Michałowska, I., Szafran, E., Bilińska, Z. T., Bobkowski, W., & Płoski, R. (2023). Double Heterozygous Pathogenic Variants in the LOX and PKD1 Genes in a 5-Year-Old Patient with Thoracic Aortic Aneurysm and Polycystic Kidney Disease. Genes, 14(11), 1983. https://doi.org/10.3390/genes14111983