


Hereditary Thrombotic Thrombocytopenic Purpura

Abstract
:1. Introduction
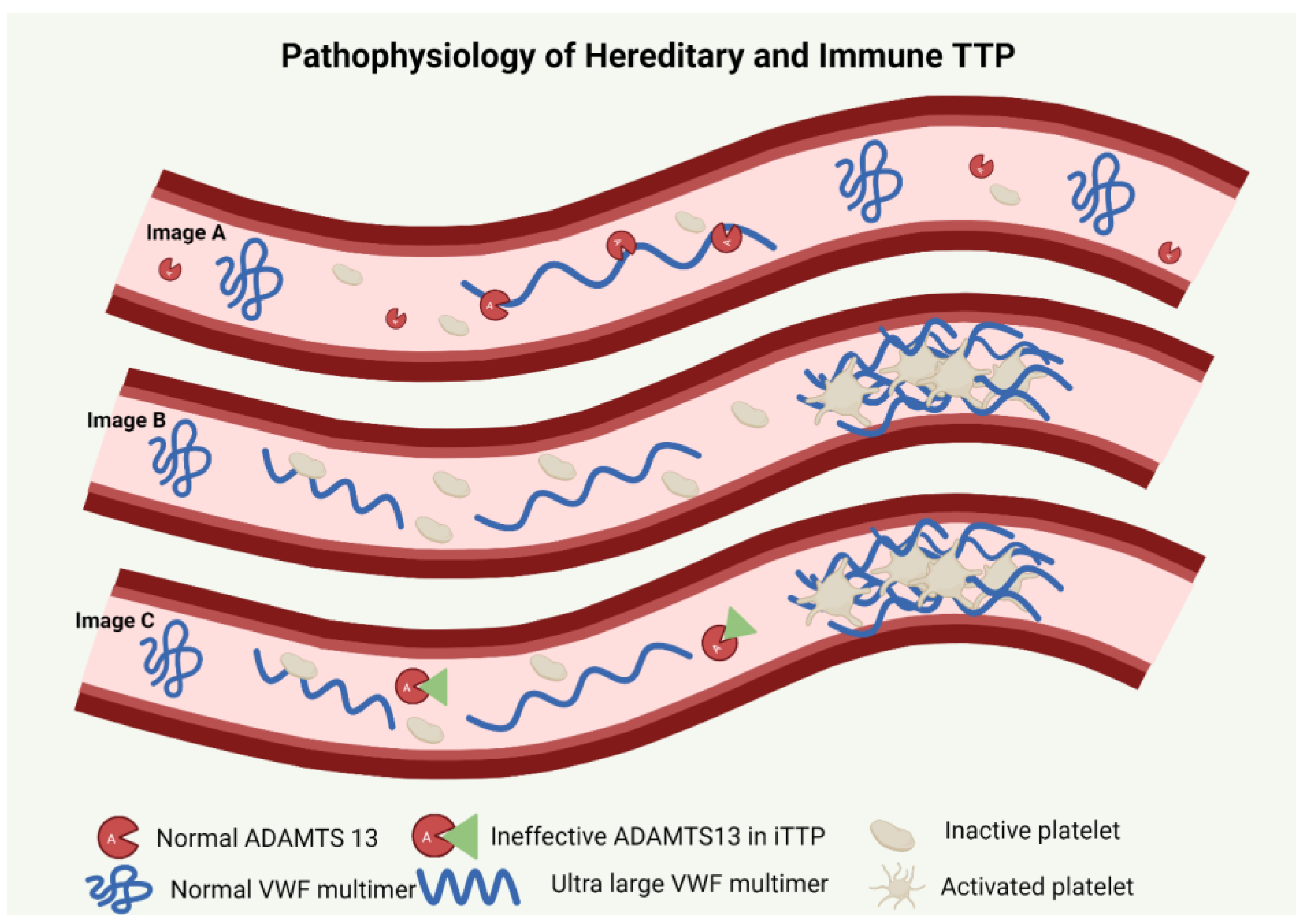
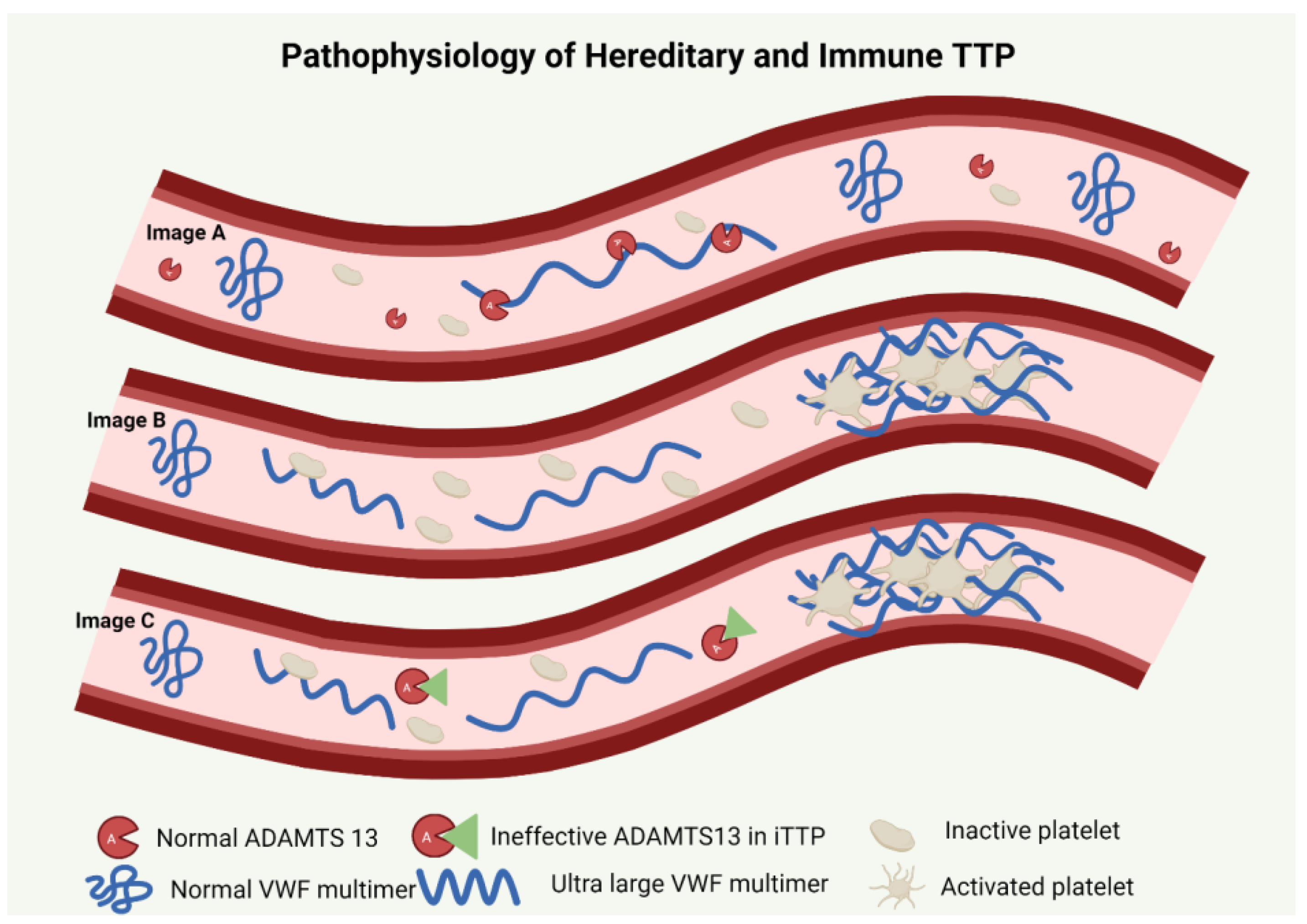
2. Pathophysiology
3. Clinical Presentation and Differential Diagnoses
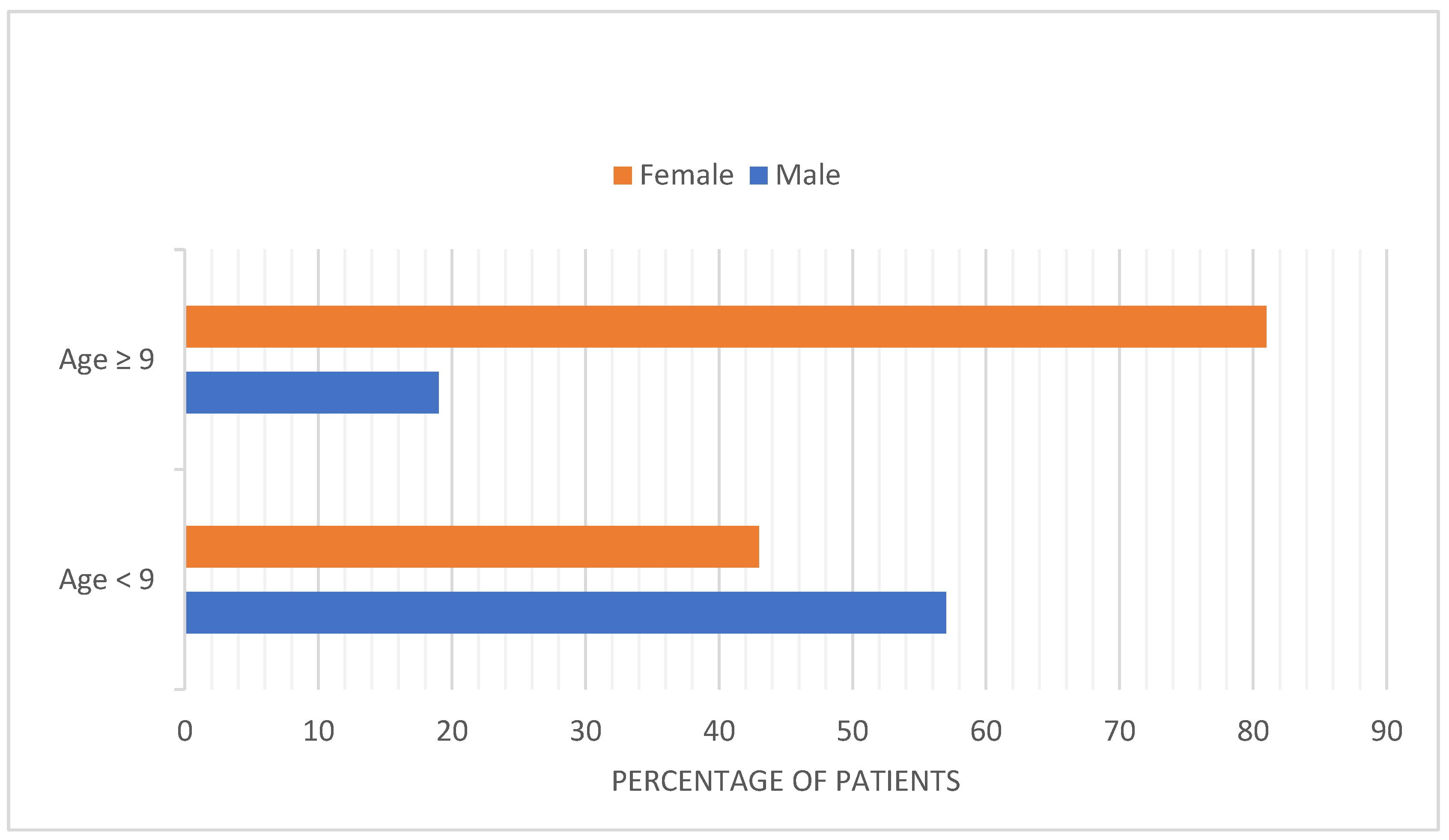
3.1. Newborn Infants
- Case 1: A Case of hTTP in a Newborn Infant [19]
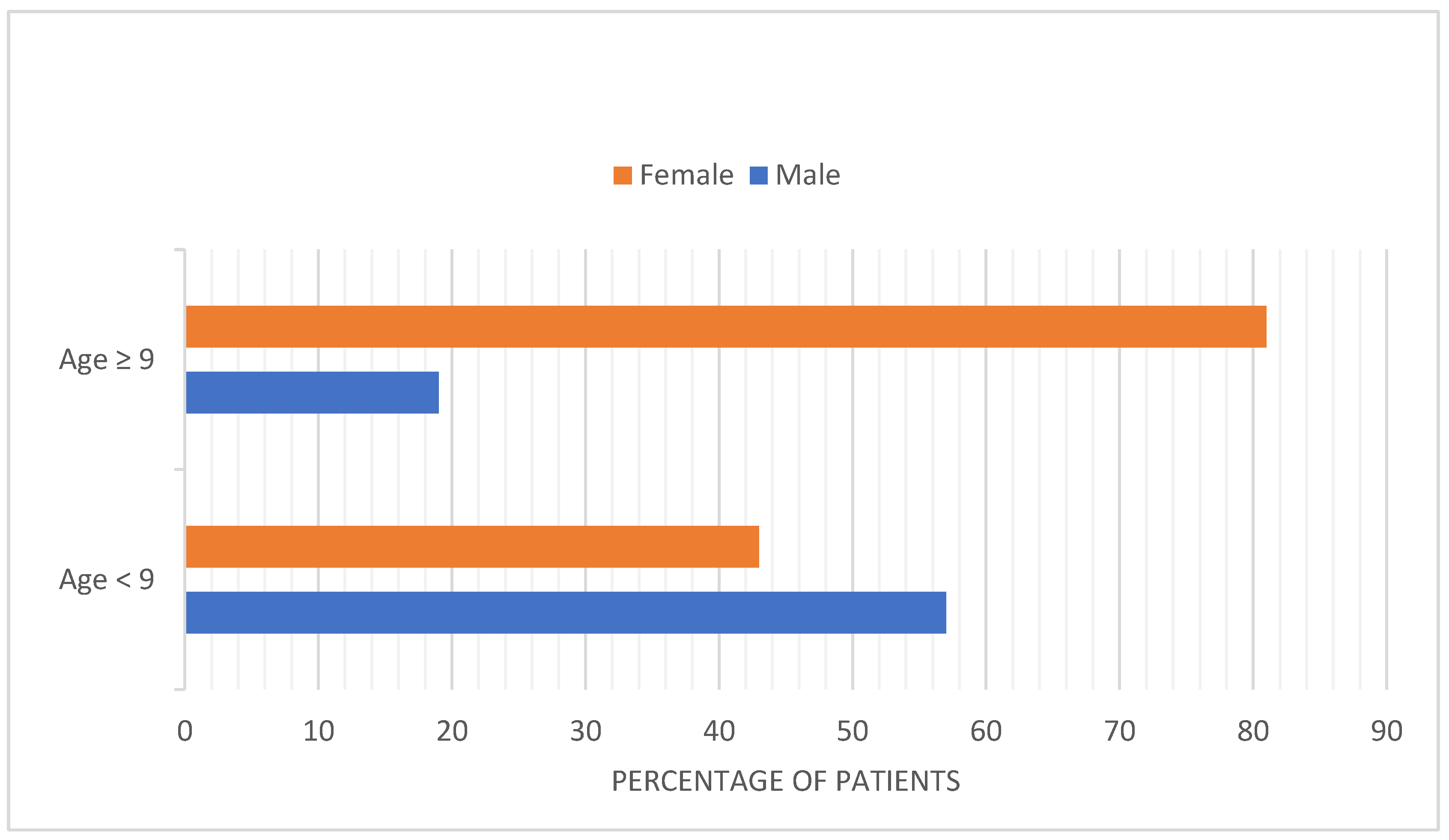
3.2. Children
- Case 2: A Case of Delayed Diagnosis of iTTP in a Child
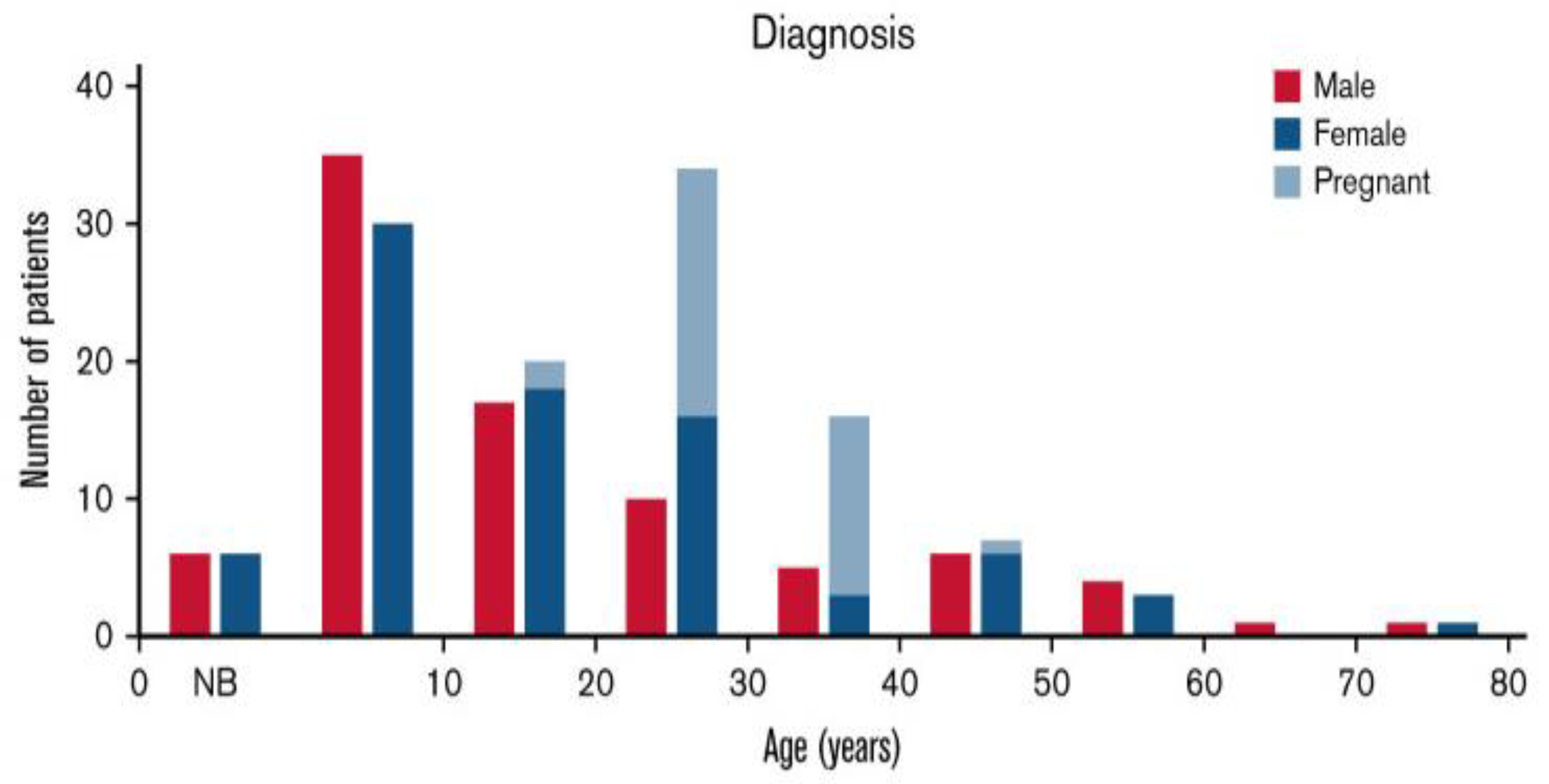
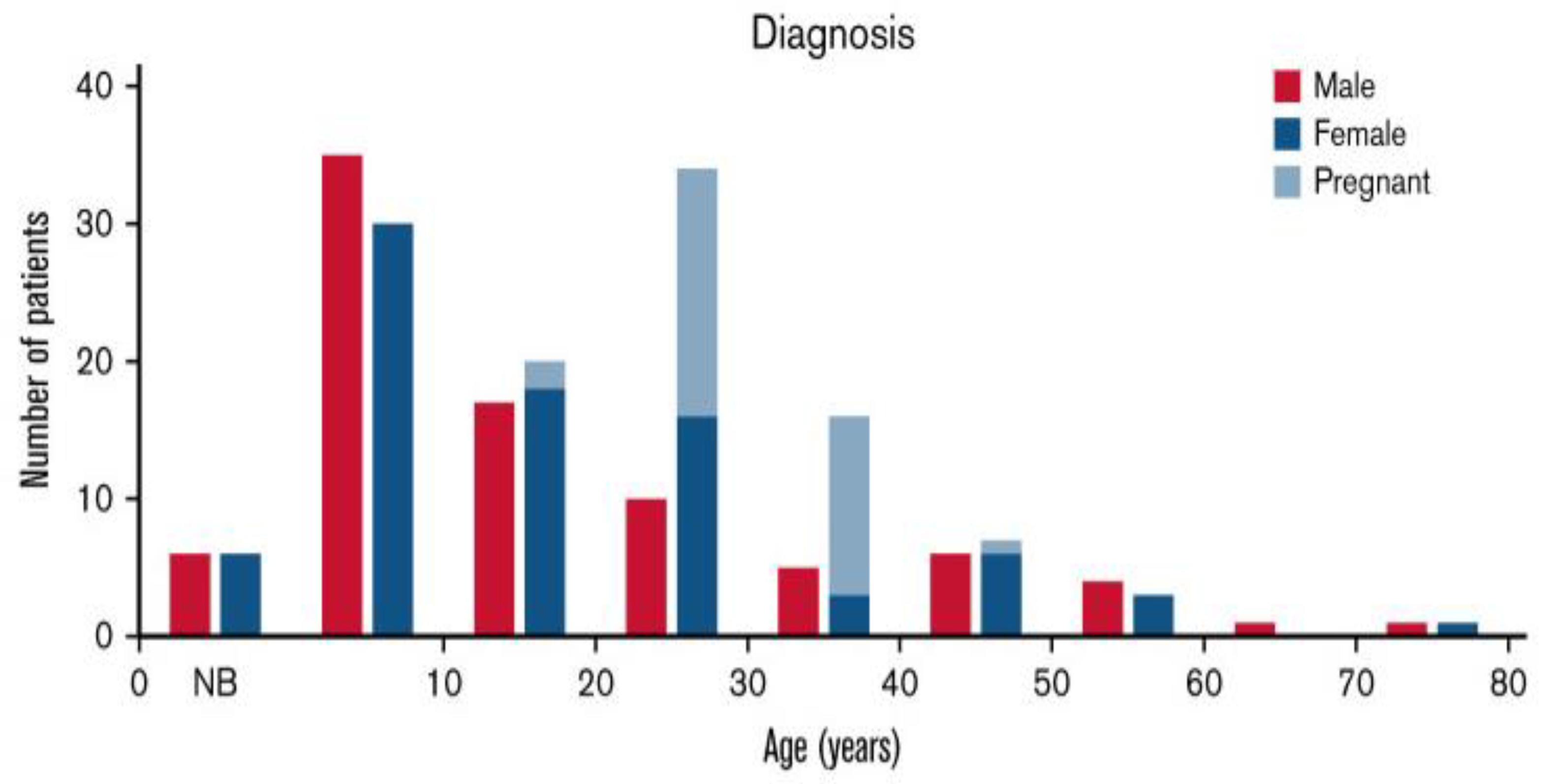
3.3. Adults
- Case 3: Pregnancy in a Woman with hTTP [37]
- Case 4: Different Clinical Features in Siblings with hTTP
4. iTTP
5. Diagnostic Evaluation
6. Management Strategies
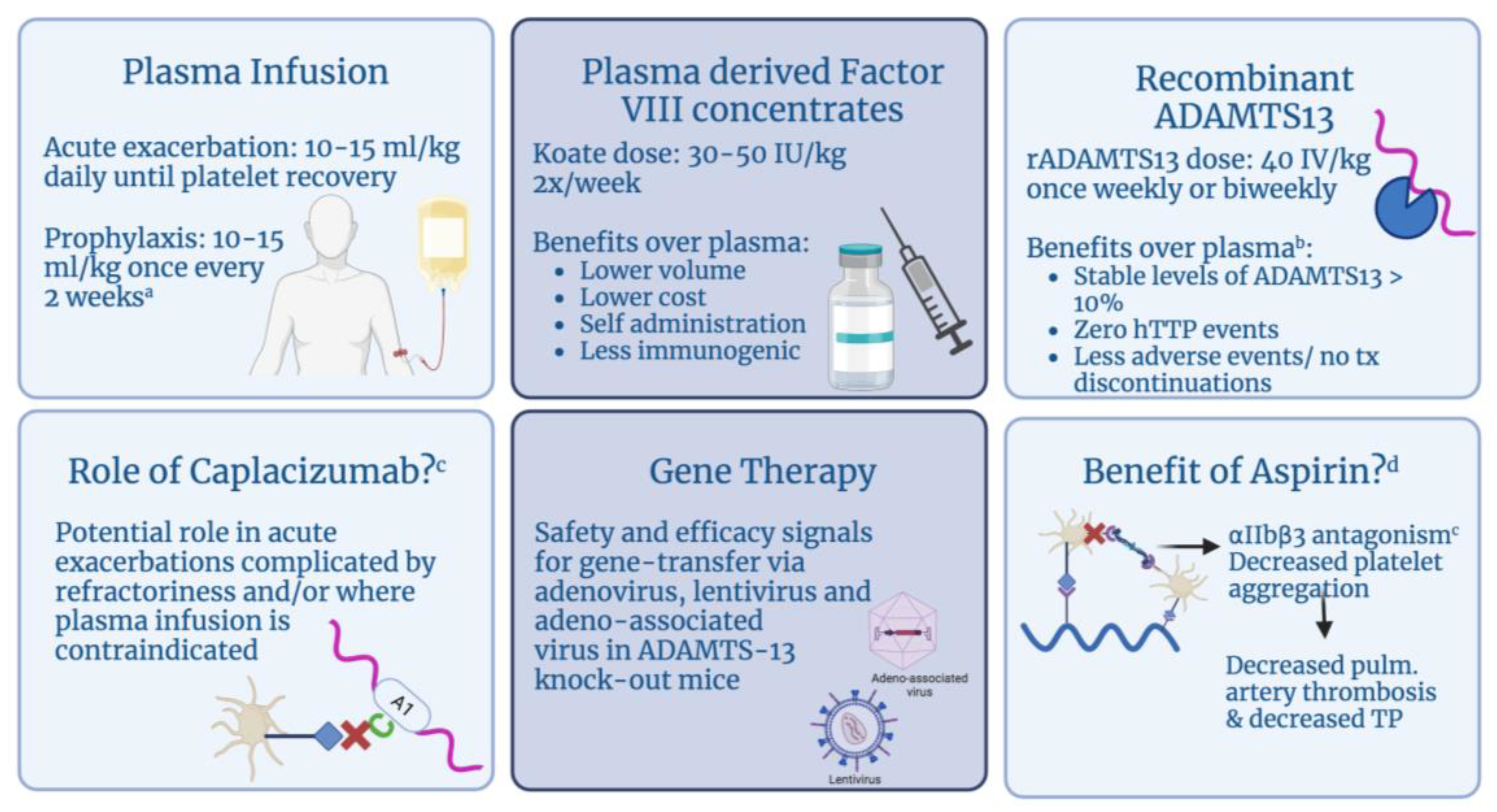
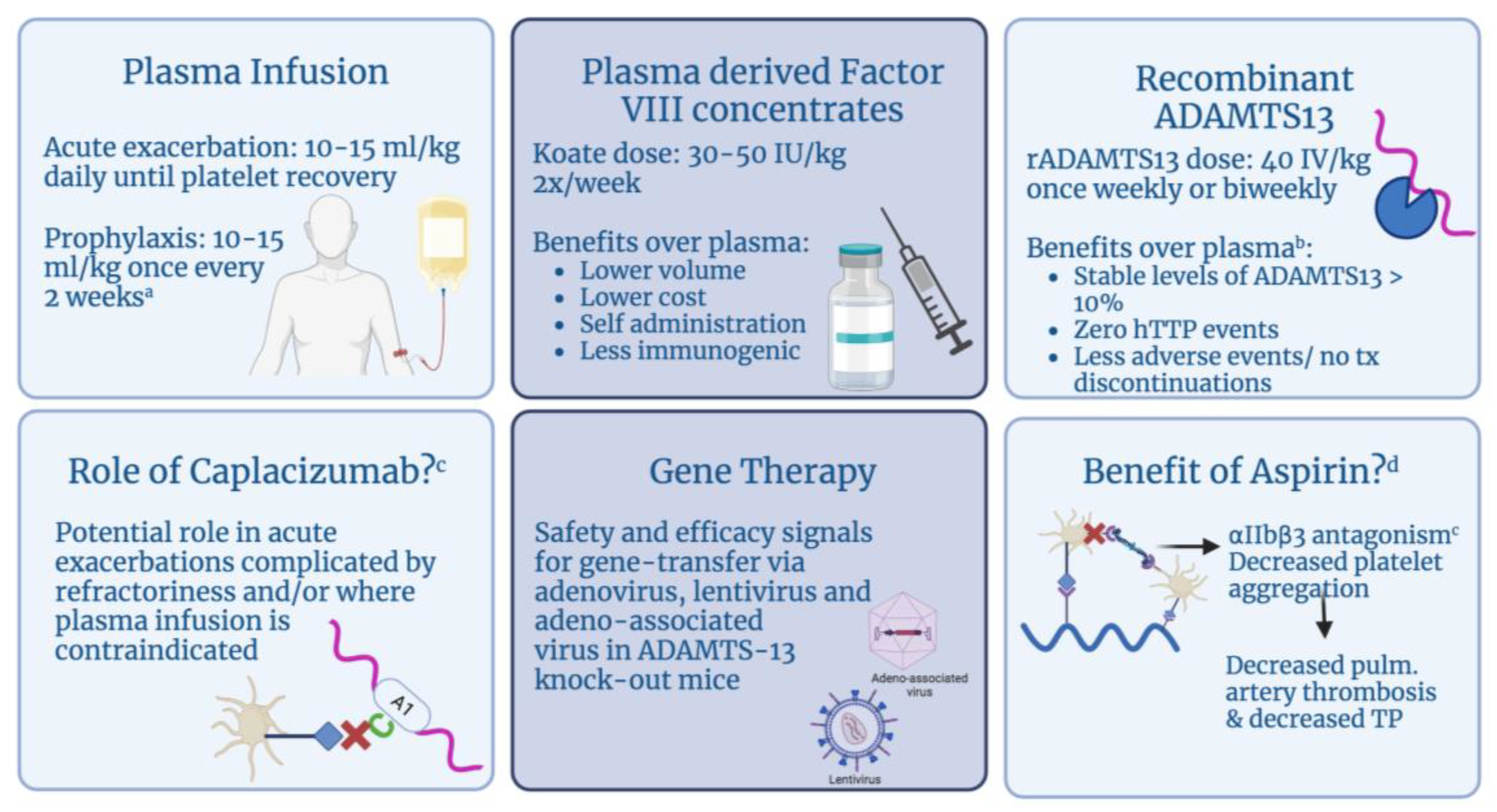
6.1. Plasma
6.2. Plasma-Derived FACTOR VIII Concentrates
6.3. Recombinant ADAMTS13
6.4. Immune-Modulating Therapies
6.5. Novel Therapies
6.6. Aspirin
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kremer Hovinga, J.A.; George, J.N. Hereditary Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 381, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [PubMed]
- South, K.; Lane, D.A. ADAMTS-13 and von Willebrand factor: A dynamic duo. J. Thromb. Haemost. 2018, 16, 6–18. [Google Scholar] [CrossRef]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. von Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, J.; Lopez, J.A. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat. Commun. 2015, 6, 7858. [Google Scholar] [CrossRef]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef]
- van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knöbl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The International Hereditary Thrombotic Thrombocytopenic Purpura Registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef]
- Kremer Hovinga, J.A.; Heeb, S.R.; Skowronska, M.; Schaller, M. Pathophysiology of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. J. Thromb. Haemost. 2018, 16, 618–629. [Google Scholar] [CrossRef]
- Lotta, L.A.; Garagiola, I.; Palla, R.; Cairo, A.; Peyvandi, F. ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum. Mutat. 2010, 31, 11–19. [Google Scholar] [CrossRef]
- Lotta, L.A.; Wu, H.M.; Mackie, I.J.; Noris, M.; Veyradier, A.; Scully, M.A.; Remuzzi, G.; Coppo, P.; Liesner, R.; Donadelli, R.; et al. Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura. Blood 2012, 120, 440–448. [Google Scholar] [CrossRef]
- Fujimura, Y.; Matsumoto, M.; Isonishi, A.; Yagi, H.; Kokame, K.; Soejima, K.; Murata, M.; Miyata, T. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 283–301. [Google Scholar] [CrossRef]
- von Krogh, A.S.; Quist-Paulsen, P.; Waage, A.; Langseth, O.; Thorstensen, K.; Brudevold, R.; Tjønnfjord, G.E.; Largiadèr, C.R.; Lämmle, B.; Hovinga, J.A.K. High prevalence of hereditary thrombotic thrombocytopenic purpura in central Norway: From clinical observation to evidence. J. Thromb. Haemost. 2016, 14, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y.; Lammle, B.; Tanabe, S.; Sakai, K.; Kimura, T.; Kokame, K.; Miyata, T.; Takahashi, Y.; Taniguchi, S.; Matsumoto, M. Patent ductus arteriosus generates neonatal hemolytic jaundice with thrombocytopenia in Upshaw-Schulman syndrome. Blood Adv. 2019, 3, 3191–3195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Li, Z.; Li, Z.; Zhang, L.; Jian, S.; Wang, C.; Song, Y.; Lv, Z.; Tang, X.; et al. Early indicators of neonatal-onset hereditary thrombotic thrombocytopenia purpura. Res. Pract. Thromb. Haemost. 2022, 6, e12820. [Google Scholar] [CrossRef] [PubMed]
- George, J.N. Hereditary thrombotic thrombocytopenic purpura: The risk for death at birth. Res. Pract. Thromb. Haemost. 2022, 6, e12840. [Google Scholar] [CrossRef]
- Borogovac, A.; Reese, J.A.; Gupta, S.; George, J.N. Morbidities and mortality in patients with hereditary thrombotic thrombocytopenic purpura. Blood Adv. 2022, 6, 750–759. [Google Scholar] [CrossRef]
- Stubbs, M.J.; Kendall, G.; Scully, M. Recombinant ADAMTS13 in Severe Neonatal Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2022, 387, 2391–2392. [Google Scholar] [CrossRef]
- Delaney, M.; Matthews, D.C. Hemolytic disease of the fetus and newborn: Managing the mother, fetus, and newborn. Hematol. Am. Soc. Hematol. Educ. Program. 2015, 2015, 146–151. [Google Scholar] [CrossRef]
- Gabbay, J.M.; Agneta, E.M.; Turkington, S.; Bajaj, B.M.; Sinha, B.; Geha, T. Rates of phototherapy among ABO-incompatible newborns with a negative direct antiglobulin test. J. Perinatol. 2023, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Y.R.; Kumar, C.G. Morbidity of ABO haemolytic disease in the newborn. Paediatr. Int. Child. Health. 2012, 32, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Journeycake, J.M.; Borogovac, A.; George, J.N. Recognizing and managing hereditary and acquired thrombotic thrombocytopenic purpura in infants and children. Pediatr. Blood Cancer. 2021, 68, e28949. [Google Scholar] [CrossRef] [PubMed]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Borogovac, A.; George, J.N. Stroke and myocardial infarction in hereditary thrombotic thrombocytopenic purpura: Similarities to sickle cell anemia. Blood Adv. 2019, 3, 3973–3976. [Google Scholar] [CrossRef]
- Alwan, F.; Mahdi, D.; Tayabali, S.; Cipolotti, L.; Lakey, G.; Hyare, H.; Scully, M. Cerebral MRI findings predict the risk of cognitive impairment in thrombotic thrombocytopenic purpura. Br. J. Haematol. 2020, 191, 868–874. [Google Scholar] [CrossRef]
- Borogovac, A.; Tarasco, E.; Kremer Hovinga, J.A.; Friedman, K.D.; Asch, A.S.; Vesely, S.K.; Prodan, C.I.; Terrell, D.R.; George, J.N. Prevalence of neuropsychiatric symptoms and stroke in patients with hereditary thrombotic thrombocytopenic purpura. Blood 2022, 140, 785–789. [Google Scholar] [CrossRef]
- George, J.N. TTP: Long-term outcomes following recovery. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 548–552. [Google Scholar] [CrossRef]
- George, J.N.; Vesely, S.K.; James, J.A. Overlapping features of thrombotic thrombocytopenic purpura and systemic lupus erythematosus. South. Med. J. 2007, 100, 512–514. [Google Scholar] [CrossRef]
- Hassan, A.; Iqbal, M.; George, J.N. Additional autoimmune disorders in patients with acquired autoimmune thrombotic thrombocytopenic purpura. Am. J. Hematol. 2019, 94, E172–E174. [Google Scholar] [CrossRef]
- Roriz, M.; Landais, M.; Desprez, J.; Barbet, C.; Azoulay, E.; Galicier, L.; Wynckel, A.; Baudel, J.L.; Provôt, F.; Pène, F.; et al. Risk Factors for Autoimmune Diseases Development After Thrombotic Thrombocytopenic Purpura. Medicine 2015, 94, e1598. [Google Scholar] [CrossRef] [PubMed]
- Tarasco, E.; Butikofer, L.; Friedman, K.D.; George, J.N.; Hrachovinova, I.V.; Knöbl, P.N.; Matsumoto, M.; von Krogh, A.S.; Aebi-Huber, I.; Cermakova, Z.; et al. Annual incidence and severity of acute episodes in hereditary thrombotic thrombocytopenic purpura. Blood 2021, 137, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Kasht, R.; Borogovac, A.; George, J.N. Frequency and severity of pregnancy complications in women with hereditary thrombotic thrombocytopenic purpura. Am. J. Hematol. 2020, 95, E316–E318. [Google Scholar] [CrossRef] [PubMed]
- Ernst, L.M. Maternal vascular malperfusion of the placental bed. APMIS 2018, 126, 551–560. [Google Scholar] [CrossRef]
- Soffer, M.D.; Bendapudi, P.K.; Roberts, D.J.; Edelson, P.K.; Kuter, D.J.; Ecker, J.L.; Bryant, A.; Goldfarb, I.T. Congenital thrombotic thrombocytopenic purpura (TTP) with placental abruption despite maternal improvement: A case report. BMC Pregnancy Childbirth 2020, 20, 365. [Google Scholar] [CrossRef]
- Perez Botero, J.; Reese, J.A.; George, J.N.; McIntosh, J.J. Severe thrombocytopenia and microangiopathic hemolytic anemia in pregnancy: A guide for the consulting hematologist. Am. J. Hematol. 2021, 96, 1655–1665. [Google Scholar] [CrossRef]
- Asmis, L.M.; Serra, A.; Krafft, A.; Licht, A.; Leisinger, E.; Henschkowski-Serra, J.; Ganter, M.T.; Hauptmann, S.; Tinguely, M.; Hovinga, J.A.K. Recombinant ADAMTS13 for Hereditary Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2022, 387, 2356–2361. [Google Scholar] [CrossRef]
- Sonneveld, M.A.; de Maat, M.P.; Portegies, M.L.; Kavousi, M.; Hofman, A.; Turecek, P.L.; Rottensteiner, H.; Scheiflinger, F.; Koudstaal, P.J.; Ikram, M.A.; et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood 2015, 126, 2739–2746. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Yu, J.; Brown, J.; Wei, A.; Selvakumar, S.; Gerber, G.F.; Moliterno, A.R.; Streiff, M.B.; Kraus, P.; Logue, C.M.; et al. Silent cerebral infarction during immune TTP remission: Prevalence, predictors, and impact on cognition. Blood 2023, 142, 325–335. [Google Scholar] [CrossRef]
- Scully, M.; Knobl, P.; Kentouche, K.; Rice, L.; Windyga, J.; Schneppenheim, R.; Kremer Hovinga, J.A.; Kajiwara, M.; Fujimura, Y.; Maggiore, C. Recombinant ADAMTS-13: First-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood 2017, 130, 2055–2063. [Google Scholar] [CrossRef]
- Shao, B.; Nusrat, S.; George, J.N.; Xia, L. Aspirin prophylaxis for hereditary and acquired thrombotic thrombocytopenic purpura? Am. J. Hematol. 2022, 97, E304–E306. [Google Scholar] [CrossRef]
- Taylor, A.; Vendramin, C.; Oosterholt, S.; Della Pasqua, O.; Scully, M. Pharmacokinetics of plasma infusion in congenital thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2019, 17, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2496–2502. [Google Scholar] [CrossRef]
- Naik, S.; Mahoney, D.H. Successful treatment of congenital TTP with a novel approach using plasma-derived factor VIII. J. Pediatr. Hematol. Oncol. 2013, 35, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Mannucci, P.M.; Valsecchi, C.; Pontiggia, S.; Farina, C.; Retzios, A.D. ADAMTS13 content in plasma-derived factor VIII/von Willebrand factor concentrates. Am. J. Hematol. 2013, 88, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Andrews, R.K. Illustrated State-of-the-Art Capsules of the ISTH 2019 Congress in Melbourne, Australia. Res. Pract. Thromb. Haemost. 2019, 3, 431–497. [Google Scholar] [CrossRef]
- Aledort, L.M.; Boggio, L.; Davis, J.A.; Gauger, C.; Kobrinsky, N.L.; Rajasekhar, A.; Shapiro, R.; Torres, M.; Ulsh, P.J. Congenital Thrombotic Thrombocytopenia Purpura—Safer Treatment with Plasma-Derived Viral-Attenuated Clotting Factor. Blood 2015, 126, 3459. [Google Scholar] [CrossRef]
- Chrisentery-Singleton, T.; Boggio, L.N.; Carcao, M.; Ibrahimi, S.; Khan, O.; Mahajerin, A.; Rajasekhar, A.; Sharma, V.; Steele, M.; Torres, M.; et al. Long-Term Follow-up of Patients with Congenital Thrombotic Thrombocytopenia Purpura Receiving Plasma-Derived Factor VIII Containing (Koate®). Blood 2022, 140 (Suppl. S1), 5644–5646. [Google Scholar] [CrossRef]
- ISTH. MS Phase 3 prospective, randomized, controlled, open-label, multicenter, crossover study of recombinant ADAMTS13 in patients with congenital thrombotic thrombocytopenic purpura; ISTH: Carrboro, NC, USA, 2023. [Google Scholar]
- Boothby, A.; Mazepa, M. Caplacizumab for congenital thrombotic thrombocytopenic purpura. Am. J. Hematol. 2022, 97, E420–E421. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hansson, E.; Delaey, B.; Callewaert, F.; De Passos Sousa, R.; Sargentini-Maier, M.L. Caplacizumab Model-Based Dosing Recommendations in Pediatric Patients with Acquired Thrombotic Thrombocytopenic Purpura. J. Clin. Pharmacol. 2022, 62, 409–421. [Google Scholar] [CrossRef]
- Dutt, T.; Shaw, R.J.; Stubbs, M.; Yong, J.; Bailiff, B.; Cranfield, T.; Crowley, M.P.; Desborough, M.J.R.; Eyre, T.A.; Gooding, R.; et al. Real-world experience with caplacizumab in the management of acute TTP. Blood 2021, 137, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Trionfini, P.; Tomasoni, S.; Galbusera, M.; Motto, D.; Longaretti, L.; Corna, D.; Remuzzi, G.; Benigni, A. Adenoviral-mediated gene transfer restores plasma ADAMTS13 antigen and activity in ADAMTS13 knockout mice. Gene Ther. 2009, 16, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Niiya, M.; Endo, M.; Shang, D.; Zoltick, P.W.; Muvarak, E.N.; Cao, W.; Jin, S.-Y.; Skipwith, C.G.; Motto, D.G.; Flake, A.W.; et al. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol. Ther. 2009, 17, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Y.; Xiao, J.; Bao, J.; Zhou, S.; Wright, J.F.; Zheng, X.L. AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood 2013, 121, 3825–3829. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector–mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef]
- Shao, B.; Hoover, C.; Shi, H.; Kondo, Y.; Lee, R.H.; Chen, J.; Shan, X.; Song, J.; McDaniel, J.M.; Zhou, M.; et al. Deletion of platelet CLEC-2 decreases GPIbα-mediated integrin αIIbβ3 activation and decreases thrombosis in TTP. Blood 2022, 139, 2523–2533. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Hereditary TTP (4 Infants) | ABO Incompatibility (20 Infants) |
|---|---|---|
| Family history | Two patients each had one older sibling who died 2 days after birth with jaundice, hemolysis | No infant deaths |
| Jaundice onset (h) | 10 (5–13) | 69 (18–82) |
| Bilirubin (mg/dl), maximum | 24 (38 h after birth) | 16 (74 h after birth) |
| Bilirubin response to phototherapy, IVIg a | 0 | 20 (100%) |
| Hemoglobin (g/dL, mean, minimum) | 10.6 | 16.3 |
| Platelet count (/μL, mean, minimum) | 17,000 | 291,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nusrat, S.; Beg, K.; Khan, O.; Sinha, A.; George, J. Hereditary Thrombotic Thrombocytopenic Purpura. Genes 2023, 14, 1956. https://doi.org/10.3390/genes14101956
Nusrat S, Beg K, Khan O, Sinha A, George J. Hereditary Thrombotic Thrombocytopenic Purpura. Genes. 2023; 14(10):1956. https://doi.org/10.3390/genes14101956
Chicago/Turabian StyleNusrat, Sanober, Kisha Beg, Osman Khan, Arpan Sinha, and James George. 2023. "Hereditary Thrombotic Thrombocytopenic Purpura" Genes 14, no. 10: 1956. https://doi.org/10.3390/genes14101956
APA StyleNusrat, S., Beg, K., Khan, O., Sinha, A., & George, J. (2023). Hereditary Thrombotic Thrombocytopenic Purpura. Genes, 14(10), 1956. https://doi.org/10.3390/genes14101956