

Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease
 , ,
, , 
Abstract
1. Introduction
2. Methods
2.1. Study Design
2.2. Sample Recruitment
2.3. TEE Diagnosis
2.4. Genomic Analysis
2.5. Sample Size Calculation
2.6. Statistical Analysis
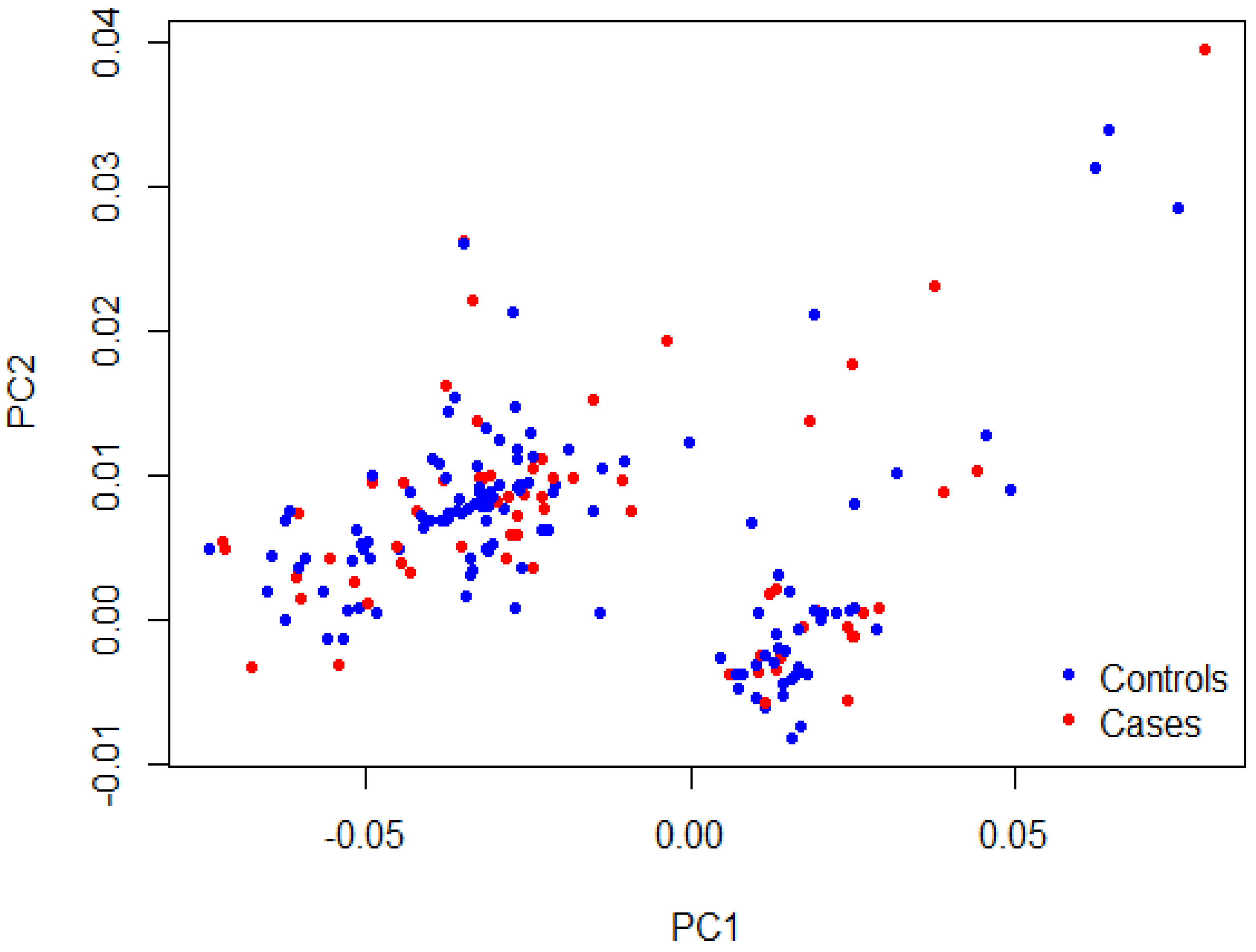
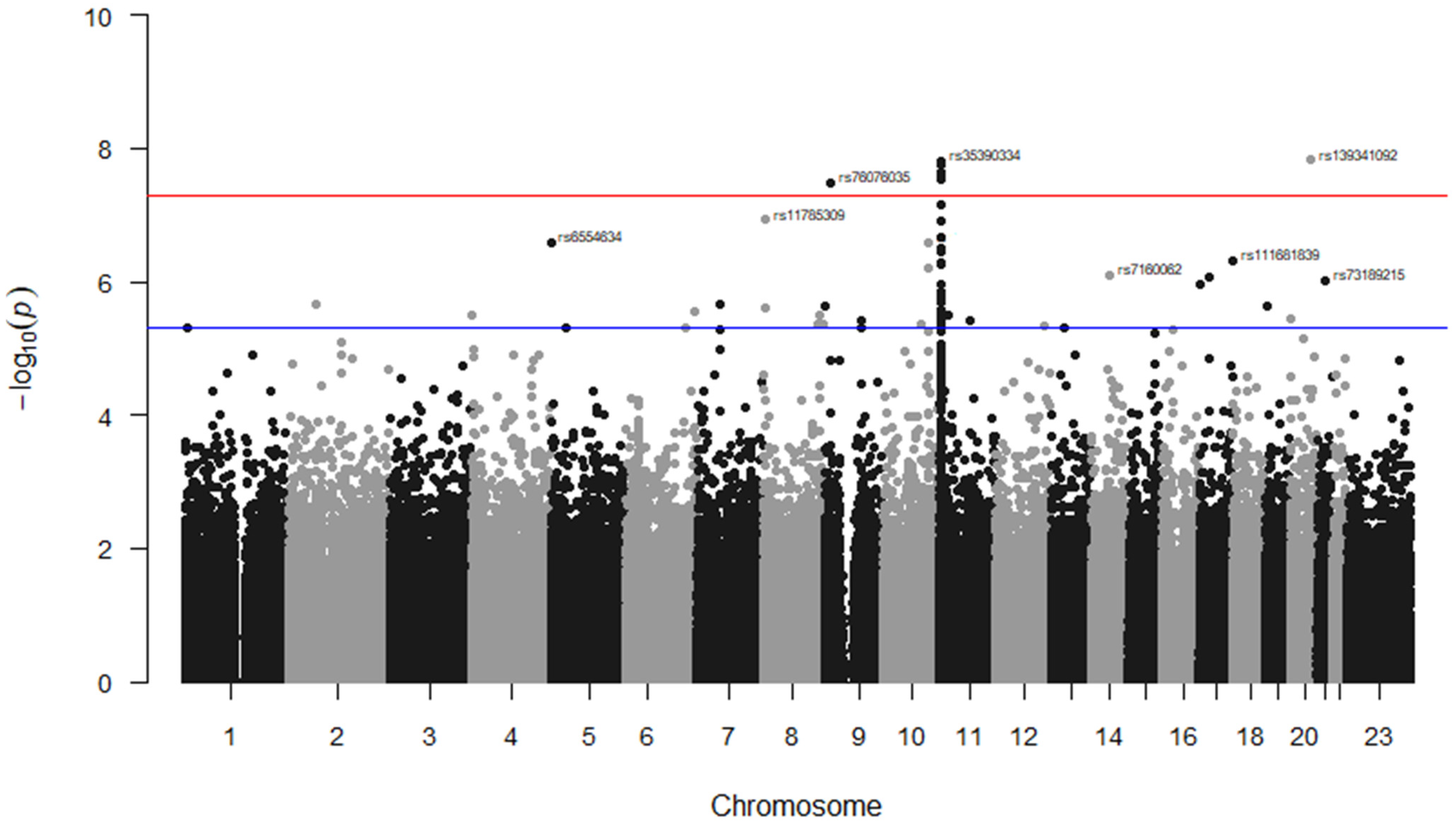
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jastaniah, W. Epidemiology of sickle cell disease in Saudi Arabia. Ann. Saudi Med. 2011, 31, 289–293. [Google Scholar] [CrossRef]
- Al-Suliman, A.; Elsarraf, N.A.; Baqishi, M.; Homrany, H.; Bousbiah, J.; Farouk, E. Patterns of mortality in adult sickle cell disease in the Al-Hasa region of Saudi Arabia. Ann. Saudi Med. 2006, 26, 487–488. [Google Scholar] [CrossRef]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-specific enzymatic amplification of β-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar] [CrossRef]
- Carden, M.A.; Fasano, R.M.; Meier, E.R. Not all red cells sickle the same: Contributions of the reticulocyte to disease pathology in sickle cell anemia. Blood Rev. 2020, 40, 100637. [Google Scholar] [CrossRef]
- Vichinsky, E. Chronic organ failure in adult sickle cell disease. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 435–439. [Google Scholar] [CrossRef]
- Janjua, T.K.; Haider, S.A.; Raza, N. Multiple complications in sickle cell anaemia. J. Pak. Med. Assoc. 2018, 68, 154–156. [Google Scholar]
- van Hamel Parsons, V.; Gardner, K.; Patel, R.; Thein, S.L. Venous thromboembolism in adults with sickle cell disease: Experience of a single centre in the UK. Ann. Hematol. 2016, 95, 227–232. [Google Scholar] [CrossRef]
- Wun, T.; Brunson, A. Sickle cell disease: An inherited thrombophilia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 640–647. [Google Scholar] [CrossRef]
- Noubouossie, D.; Key, N.S.; Ataga, K.I. Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. 2016, 30, 245–256. [Google Scholar] [CrossRef]
- Shet, A.S.; Lizarralde-Iragorri, M.A.; Naik, R.P. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica 2020, 105, 2368. [Google Scholar] [CrossRef]
- Austin, H.; Key, N.S.; Benson, J.M.; Lally, C.; Dowling, N.F.; Whitsett, C.; Hooper, W.C. Sickle cell trait and the risk of venous thromboembolism among blacks. Blood 2007, 110, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Little, I.; Vinogradova, Y.; Orton, E.; Kai, J.; Qureshi, N. Venous thromboembolism in adults screened for sickle cell trait: A population-based cohort study with nested case-control analysis. BMJ Open 2017, 7, e012665. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Beemath, A.; Meyers, F.A.; Skaf, E.; Olson, R.E. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am. J. Med. 2006, 119, 897.e7–897.e11. [Google Scholar] [CrossRef] [PubMed]
- Ohene-Frempong, K.; Weiner, S.J.; Sleeper, L.A.; Miller, S.T.; Embury, S.; Moohr, J.W.; Wethers, D.L.; Pegelow, C.H.; Gill, F.M.; The Cooperative Study of Sickle Cell Disease. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood 1998, 91, 288–294. [Google Scholar] [PubMed]
- Kirkham, F.J.; Lagunju, I.A. Epidemiology of stroke in sickle cell disease. J. Clin. Med. 2021, 10, 4232. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.P.; Streiff, M.B.; Haywood, C., Jr.; Segal, J.B.; Lanzkron, S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J. Thromb. Haemost. 2014, 12, 2010–2016. [Google Scholar] [CrossRef]
- Brunson, A.; Lei, A.; Rosenberg, A.S.; White, R.H.; Keegan, T.; Wun, T. Increased incidence of VTE in sickle cell disease patients: Risk factors, recurrence and impact on mortality. Br. J. Haematol. 2017, 178, 319–326. [Google Scholar] [CrossRef]
- Alsultan, A.; Aleem, A.; Ghabbour, H.; AlGahtani, F.H.; Al-Shehri, A.; Osman, M.E.; Kurban, K.; Alsultan, M.S.; Bahakim, H.; Al-Momen, A.M. Sickle cell disease subphenotypes in patients from Southwestern Province of Saudi Arabia. J. Pediatr. Hematol. Oncol. 2012, 34, 79–84. [Google Scholar] [CrossRef]
- Al-Gahtani, F.H. Thromboembolic Events among Patients with Sickle Cell Disease: Risk Factors and Prevalence in a Tertiary Hospital in Saudi Arabia. J. Hematol. Thrombo. Dis. 2016, 4, 1000254. [Google Scholar] [CrossRef]
- Cushman, M. Epidemiology and risk factors for venous thrombosis. Semin. Hematol. 2007, 44, 62–69. [Google Scholar] [CrossRef]
- Porter, B.; Key, N.S.; Jauk, V.C.; Adam, S.; Biggio, J.; Tita, A. Impact of sickle hemoglobinopathies on pregnancy-related venous thromboembolism. Am. J. Perinatol. 2014, 31, 805–809. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Lanzkron, S. Sickle cell disease and venous thromboembolism: What the anticoagulation expert needs to know. J. Thromb. Thrombolysis 2013, 35, 352–358. [Google Scholar] [CrossRef]
- Wu, O.; Robertson, L.; Twaddle, S.; Lowe, G.D.; Clark, P.; Greaves, M.; Walker, I.D.; Langhorne, P.; Brenkel, I.; Regan, L.; et al. Screening for thrombophilia in high-risk situations: Systematic review and cost-effectiveness analysis. The Thrombosis: Risk and Economic Assessment of Thrombophilia Screening (TREATS) study. Health Technol. Assess. 2006, 10, 1–110. [Google Scholar] [CrossRef]
- Wuthrich, R.P.; Cicvara-Muzar, S.; Booy, C.; Maly, F.E. Heterozygosity for the factor V Leiden (G1691A) mutation predisposes renal transplant recipients to thrombotic complications and graft loss. Transplantation 2001, 72, 549–550. [Google Scholar] [CrossRef]
- Al-Allawi, N.A.; Badi, A.I.; Goran, M.A.; Nerweyi, F.F.; Ballo, H.M.; Al-Mzury, N.T. The Contributions of Thrombophilic Mutations to Genetic Susceptibility to Deep Venous Thrombosis in Iraqi Patients. Genet. Test. Mol. Biomark. 2015, 19, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Konecny, F. Inherited trombophilic states and pulmonary embolism. J. Res. Med. Sci. 2009, 14, 43–56. [Google Scholar] [PubMed]
- Li, C.; Ren, H.; Chen, H.; Song, J.; Li, S.; Lee, C.; Liu, J.; Cui, Y. Prothrombin G20210A (rs1799963) polymorphism increases myocardial infarction risk in an age-related manner: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 13550. [Google Scholar] [CrossRef] [PubMed]
- Gawish, G. The Prevalence of Inherited Thrombophilic Polymorphisms in Saudi Females with Recurrent Pregnancy Loss Confirmed using Different Screening Protocols of PCR. J. Mol. Genet. Med. 2015, 9, 1747. [Google Scholar]
- Badawy, A.; AlSel, B.A.; Fawzy, M. Factor V Leiden G1691A and Prothrombin G20210A mutations are associated with repeated spontaneous miscarriage in Northern area of Saudi Arabia. Gen. Mol. Res. 2017, 16, gmr16039810. [Google Scholar] [CrossRef]
- Shafia, S.; Zargar, M.H.; Khan, N.; Ahmad, R.; Shah, Z.A.; Asimi, R. High prevalence of factor V Leiden and prothrombin G20101A mutations in Kashmiri patients with venous thromboembolism. Gene 2018, 654, 1–9. [Google Scholar] [CrossRef]
- Ehsani, M.; Imani, A.; Moravveji, A. Prevalence of factor V leiden, MTHFR C677T and MTHFR A1298C polymorphisms in patients with deep vein thrombosis in Central Iran. Mol. Biol. Rep. 2018, 45, 621–624. [Google Scholar] [CrossRef]
- Hosseini, S.; Kalantar, E.; Hosseini, M.S.; Tabibian, S.; Shamsizadeh, M.; Dorgalaleh, A. Genetic risk factors in patients with deep venous thrombosis, a retrospective case control study on Iranian population. Thromb. J. 2015, 13, 35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghaznavi, H.; Soheili, Z.; Samiei, S.; Soltanpour, M.S. Association of Methylenetetrahydrofolate Reductase C677T Polymorphism with Hyperhomocysteinemia and Deep Vein Thrombosis in the Iranian Population. Vasc. Spec. Int. 2015, 31, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kamei, H.; Nakamura, T.; Nagai, S.; Ishigami, M.; Hamajima, N. Possible Association Between the Methylenetetrahydrofolate Reductase Gene C677T Polymorphism and Preexisting Portal Vein Thrombosis in Liver Transplant Recipients. Exp. Clin. Transplant. 2016, 14, 313–316. [Google Scholar] [PubMed]
- Ventura, P.; Venturelli, G.; Marcacci, M.; Fiorini, M.; Marchini, S.; Cuoghi, C.; Pietrangelo, A. Hyperhomocysteinemia and MTHFR C677T polymorphism in patients with portal vein thrombosis complicating liver cirrhosis. Thromb. Res. 2016, 141, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Spronk, K.J.; Olivero, A.D.; Haw, M.P.; Vettukattil, J.J. Methylenetetrahydrofolate Reductase C677T: Hypoplastic Left Heart and Thrombosis. World J. Pediatr. Congenit. Heart Surg. 2015, 6, 643–645. [Google Scholar] [CrossRef]
- Garakanidze, S.; Costa, E.; Bronze-Rocha, E.; Santos-Silva, A.; Nikolaishvili, G.; Nakashidze, I.; Kakauridze, N.; Glonti, S.; Khukhunaishvili, R.; Koridze, M.; et al. Methylenetetrahydrofolate Reductase Gene Polymorphism (C677T) as a Risk Factor for Arterial Thrombosis in Georgian Patients. Clin. Appl. Thromb. Hemost. 2018, 24, 1061–1066. [Google Scholar] [CrossRef]
- Settin, A.A.; Alghasham, A.; Ali, A.; Dowaidar, M.; Ismail, H. Frequency of thrombophilic genetic polymorphisms among Saudi subjects compared with other populations. Hematology 2012, 17, 176–182. [Google Scholar] [CrossRef]
- Pandey, S.K.; Meena, A.; Kishor, K.; Mishra, R.M.; Pandey, S.; Saxena, R. Prevalence of factor V Leiden G1691A, MTHFR C677T, and prothrombin G20210A among Asian Indian sickle cell patients. Clin. Appl. Thromb. Hemost. 2012, 18, 320–323. [Google Scholar] [CrossRef]
- Nishank, S.S.; Singh, M.P.; Yadav, R. Clinical impact of factor V Leiden, prothrombin G20210A, and MTHFR C677T mutations among sickle cell disease patients of Central India. Eur. J. Haematol. 2013, 91, 462–466. [Google Scholar] [CrossRef]
- Kangne, H.K.; Jijina, F.F.; Italia, Y.M.; Jain, D.L.; Nadkarni, A.H.; Ghosh, K.K.; Colah, R.B. The Prevalence of Factor V Leiden (G1691A) and Methylenetetrahydrofolate Reductase C677T Mutations in Sickle Cell Disease in Western India. Clin. Appl. Thromb. Hemost. 2015, 21, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Moreira Neto, F.; Lourenco, D.M.; Noguti, M.A.; Morelli, V.M.; Gil, I.C.; Beltrao, A.C.; Figueiredo, M.S. The clinical impact of MTHFR polymorphism on the vascular complications of sickle cell disease. Braz. J. Med. Biol. Res. 2006, 39, 1291–1295. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hatzlhofer, B.L.; Bezerra, M.A.; Santos, M.N.; Albuquerque, D.M.; Freitas, E.M.; Costa, F.F.; Araujo, A.S.; Muniz, M.T. MTHFR polymorphic variant C677T is associated to vascular complications in sickle-cell disease. Genet. Test. Mol. Biomark. 2012, 16, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Nefissi, R.B.; Ouali, F.; Massaoud, T.; Gritli, N. Thrombophilic Mutations among Patients with Sickle Cell Disease. Clin. Lab. 2017, 63, 1815–1818. [Google Scholar] [CrossRef]
- Fawaz, N.A.; Bashawery, L.; Al-Sheikh, I.; Qatari, A.; Al-Othman, S.S.; Almawi, W.Y. Factor V-Leiden, prothrombin G20210A, and MTHFR C677T mutations among patients with sickle cell disease in Eastern Saudi Arabia. Am. J. Hematol. 2004, 76, 307–309. [Google Scholar] [CrossRef]
- Shaikho, E.M.; Farrell, J.J.; Alsultan, A.; Qutub, H.; Al-Ali, A.K.; Figueiredo, M.S.; Chui, D.H.K.; Farrer, L.A.; Murphy, G.J.; Mostoslavsky, G.; et al. A phased SNP-based classification of sickle cell anemia HBB haplotypes. BMC Genom. 2017, 18, 608. [Google Scholar] [CrossRef]
- Witte, J.S.; Elston, R.C.; Schork, N.J. Genetic dissection of complex traits. Nat. Genet. 1996, 12, 355–356. [Google Scholar] [CrossRef]
- Eliades, N.-G.; Eliades, D.G. HAPLOTYPE ANALYSIS: Software for analysis of haplotype data. In Forest Genetics and Forest Tree Breeding; Georg-August University: Goettingen, Germany, 2009. [Google Scholar]
- Greliche, N.; Germain, M.; Lambert, J.-C.; Cohen, W.; Bertrand, M.; Dupuis, A.-M.; Letenneur, L.; Lathrop, M.; Amouyel, P.; Morange, P.-E. A genome-wide search for common SNP x SNP interactions on the risk of venous thrombosis. BMC Med. Genet. 2013, 14, 36. [Google Scholar] [CrossRef]
- Heit, J.A.; Armasu, S.M.; McCauley, B.M.; Kullo, I.J.; Sicotte, H.; Pathak, J.; Chute, C.G.; Gottesman, O.; Bottinger, E.P.; Denny, J.C. Identification of unique venous thromboembolism-susceptibility variants in African-Americans. Thromb. Haemost. 2017, 117, 758–768. [Google Scholar] [CrossRef]
- Lindström, S.; Wang, L.; Smith, E.N.; Gordon, W.; van Hylckama Vlieg, A.; De Andrade, M.; Brody, J.A.; Pattee, J.W.; Haessler, J.; Brumpton, B.M. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood 2019, 134, 1645–1657. [Google Scholar] [CrossRef]
- Arning, A.; Hiersche, M.; Witten, A.; Kurlemann, G.; Kurnik, K.; Manner, D.; Stoll, M.; Nowak-Göttl, U. A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood 2012, 120, 5231–5236. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R.P.; Vieira, C.; Adanho, C.S.A.; Guarda, C.C.; Santana, S.S.; Figueiredo, C.V.B.; Aleluia, M.M.; Pitanga, T.N.; Fiuza, L.M.; Maffili, V.V. Genome Wide Association Study of Sickle Cell Disease Individuals with Stroke Risk. Blood 2016, 128, 4852. [Google Scholar] [CrossRef]
- Dichgans, M.; Malik, R.; König, I.R.; Rosand, J.; Clarke, R.; Gretarsdottir, S.; Thorleifsson, G.; Mitchell, B.D.; Assimes, T.L.; Levi, C. Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome-wide analysis of common variants. Stroke 2014, 45, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.A.; Seshadri, S.; Bis, J.C.; Fornage, M.; DeStefano, A.L.; Aulchenko, Y.S.; Debette, S.; Lumley, T.; Folsom, A.R.; Van Den Herik, E.G. Genomewide association studies of stroke. N. Engl. J. Med. 2009, 360, 1718–1728. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Holm, H.; Gretarsdottir, S.; Thorleifsson, G.; Walters, G.B.; Thorgeirsson, G.; Gulcher, J.; Mathiesen, E.B.; Njølstad, I.; Nyrnes, A. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 2009, 41, 876–878. [Google Scholar] [CrossRef]
- Holliday, E.G.; Maguire, J.M.; Evans, T.-J.; Koblar, S.A.; Jannes, J.; Sturm, J.W.; Hankey, G.J.; Baker, R.; Golledge, J.; Parsons, M.W. Common variants at 6p21. 1 are associated with large artery atherosclerotic stroke. Nat. Genet. 2012, 44, 1147–1151. [Google Scholar] [CrossRef]
- Zholdybayeva, E.V.; Medetov, Y.Z.; Aitkulova, A.M.; Makhambetov, Y.T.; Akshulakov, S.K.; Kaliyev, A.B.; Talzhanov, Y.A.; Kulmambetova, G.N.; Iskakova, A.N.; Ramankulov, Y.M. Genetic risk factors for intracranial aneurysm in the Kazakh population. J. Mol. Neurosci. 2018, 66, 135–145. [Google Scholar] [CrossRef]
- Sebastiani, P.; Ramoni, M.F.; Nolan, V.; Baldwin, C.T.; Steinberg, M.H. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat. Genet. 2005, 37, 435–440. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Frohlich, D.M.; Howard, T.A.; Schultz, W.H.; Driscoll, C.; Nagasubramanian, R.; Mortier, N.A.; Kimble, A.C.; Aygun, B.; Adams, R.J. Genetic predictors for stroke in children with sickle cell anemia. Blood 2011, 117, 6681–6684. [Google Scholar] [CrossRef]
- Daly, A. Genetic Basis of Drug-Induced Liver Injury Linked to Commonly Prescribed Drugs. In Proceedings of the Abstracts of the XXIX International Congress of the European Association of Poison Centres and Clinical Toxicologists, Stockholm, Sweden, 12–15 May 2009; Volume 47, p. 438. [Google Scholar]
- Witte, J.S. Genome-wide association studies and beyond. Annu. Rev. Public Health 2010, 31, 9–20. [Google Scholar] [CrossRef]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R. HLA-B* 5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Bhatnagar, P.; Bean, C.J.; Steinberg, M.H.; Milton, J.N.; Casella, J.F.; Barron-Casella, E.; Arking, D.E.; DeBaun, M.R. Genome-wide association study to identify variants associated with acute severe vaso-occlusive pain in sickle cell anemia. Blood 2017, 130, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, P.; Purvis, S.; Barron-Casella, E.; DeBaun, M.R.; Casella, J.F.; Arking, D.E.; Keefer, J.R. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J. Hum. Genet. 2011, 56, 316–323. [Google Scholar] [CrossRef]
- Mtatiro, S.N.; Singh, T.; Rooks, H.; Mgaya, J.; Mariki, H.; Soka, D.; Mmbando, B.; Msaki, E.; Kolder, I.; Thein, S.L. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PLoS ONE 2014, 9, e111464. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.A.; Griffiths-Jones, S.; Pink, R.C.; Carter, D.R. Pseudogenes as regulators of biological function. Essays Biochem. 2013, 54, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ibn-Salem, J.; Muro, E.M.; Andrade-Navarro, M.A. Co-regulation of paralog genes in the three-dimensional chromatin architecture. Nucleic Acids Res. 2017, 45, 81–91. [Google Scholar] [CrossRef]
- Roth, J.R. Frameshift mutations. Annu. Rev. Genet. 1974, 8, 319–346. [Google Scholar] [CrossRef]
- Solovieff, N.; Milton, J.N.; Hartley, S.W.; Sherva, R.; Sebastiani, P.; Dworkis, D.A.; Klings, E.S.; Farrer, L.A.; Garrett, M.E.; Ashley-Koch, A. Fetal hemoglobin in sickle cell anemia: Genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood 2010, 115, 1815–1822. [Google Scholar] [CrossRef]
- Menzel, S.; Garner, C.; Rooks, H.; Spector, T.D.; Thein, S.L. H b A 2 levels in normal adults are influenced by two distinct genetic mechanisms. Br. J. Haematol. 2013, 160, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [PubMed]
- Hou, C.; Dale, R.; Dean, A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. USA 2010, 107, 3651–3656. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar]
- Morrell, C.N.; Mix, D.; Aggarwal, A.; Bhandari, R.; Godwin, M.; Owens, P.; Lyden, S.P.; Doyle, A.; Krauel, K.; Rondina, M.T. Platelet olfactory receptor activation limits platelet reactivity and growth of aortic aneurysms. J. Clin. Investig. 2022, 132, e152373. [Google Scholar] [CrossRef]
- Bosch, F.; van Dijk, F.; Briedé, S.; Groen, J.; Hanna-Sawires, R.; Klok, F.; Kaasjager, H.; Brosens, L.; Molenaar, I.; Bonsing, B.; et al. Tumor Gene Expression Associates with Venous Thromboembolism in Patients with Pancreatic Cancer. Res. Pract. Thromb. Haemost. 2021, 5 (Suppl. S2). Available online: https://abstracts.isth.org/abstract/tumor-gene-expression-associates-with-venous-thromboembolism-in-patients-with-pancreatic-cancer/ (accessed on 7 February 2023).
- Feng, J.; Zhang, Z.; Wu, X.; Mao, A.; Chang, F.; Deng, X.; Gao, H.; Ouyang, C.; Dery, K.J.; Le, K. Discovery of potential new gene variants and inflammatory cytokine associations with fibromyalgia syndrome by whole exome sequencing. PLoS ONE 2013, 8, e65033. [Google Scholar] [CrossRef]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T. A genome-wide association identified the common genetic variants influence disease severity in β 0-thalassemia/hemoglobin E. Hum. Genet. 2010, 127, 303–314. [Google Scholar] [CrossRef]
- Lee, C.-J.; Chen, T.-H.; Lim, A.M.W.; Chang, C.-C.; Sie, J.-J.; Chen, P.-L.; Chang, S.-W.; Wu, S.-J.; Hsu, C.-L.; Hsieh, A.-R. Phenome-wide analysis of Taiwan Biobank reveals novel glycemia-related loci and genetic risks for diabetes. Commun. Biol. 2022, 5, 1175. [Google Scholar] [CrossRef]
- Huang, P.; Keller, C.A.; Giardine, B.; Grevet, J.D.; Davies, J.O.; Hughes, J.R.; Kurita, R.; Nakamura, Y.; Hardison, R.C.; Blobel, G.A. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 2017, 31, 1704–1713. [Google Scholar] [CrossRef]
- Palstra, R.-J.T.S. Spatial Organisation of the [β]-globin Locus: The Active Chromatin Hub. Ph.D. Thesis, Erasmus University Rotterdam, Rotterdam, The Netherlands, 2005. [Google Scholar]
- Wormald, S.; Milla, L.; O’Connor, L. Association of candidate single nucleotide polymorphisms with somatic mutation of the epidermal growth factor receptor pathway. BMC Med. Genom. 2013, 6, 43. [Google Scholar] [CrossRef]
- Al-Ali, A.K.; Alsulaiman, A.; Alfarhan, M.; Safaya, S.; Vatte, C.B.; Albuali, W.M.; Qutub, H.O.; Alzahrani, A.J.; Milton, J.N.; Steinberg, M.H. Sickle cell disease in the Eastern Province of Saudi Arabia: Clinical and laboratory features. Am. J. Hematol. 2021, 96, E117–E121. [Google Scholar] [CrossRef] [PubMed]
- Abou-Elew, H.H.; Youssry, I.; Hefny, S.; Hashem, R.H.; Fouad, N.; Zayed, R.A. βS globin gene haplotype and the stroke risk among Egyptian children with sickle cell disease. Hematology 2018, 23, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and prediction: Lessons and challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Morova, T.; Ding, Y.; Huang, C.-C.F.; Sar, F.; Schwarz, T.; Giambartolomei, C.; Baca, S.C.; Grishin, D.; Hach, F.; Gusev, A. Optimized high-throughput screening of non-coding variants identified from genome-wide association studies. Nucleic Acids Res. 2022, 51, e18. [Google Scholar]
- Cano-Gamez, E.; Trynka, G. From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| MAF Reported by Fawaz et al. [45] | FVL (rs6025) | MTHFR (rs1801133) |
|---|---|---|
| Controls | 0.01 | 0.14 |
| Cases | 0.04 | 0.25 |
| Sample size with 80% power | 337 | 222 |
| Variables | TEE Cases, n = 65 (%) | No TEE Controls; n = 285 (%) | p-Value |
|---|---|---|---|
| Males/Females | 32/33 | 162/123 | 0.27 |
| Age in years, Mean ± SD | 35.7 ± 9.8 | 34.4 ± 10.3 | 0.35 |
| Fetal hemoglobin (HbF), Mean ± SD | 10.7 ± 8.0 | 14.1 ± 7.6 | 0.002 |
| Southwestern province | 51 (78.5) | 133 (46.7) | |
| Eastern province | 14 (21.5) | 152 (53.3) | |
| DVT only | 11 (16.9) | ||
| PE only | 18 (27.7) | ||
| Stroke only | 22 (33.8) | ||
| DVT and PE | 5 (7.7) | ||
| DVT and stroke | 3 (4.6) | ||
| PE and stroke | 2 (3.1) | ||
| Other TEEs affected different organs (eye, ear, and liver) | 4 (6.2) |
| SCD Cases with TEEs (n = 65) | SCD Controls without TEEs (n = 285) | ||
|---|---|---|---|
| Gene, Variant (Alleles) | Allele Frequencies (MAF%) | p-Value | |
| Known thrombotic gene | |||
| MTHFR, rs1801133 (A/G) | 21/109 (0.16) | 85/481 (0.15) | 0.74 |
| Previously reported with TEEs | |||
| ITPR3, rs2229637 (A/G) | 24/106 (0.18) | 166/404 (0.29) | 0.014 |
| LINC02651-RPL5P26, rs10998957 (C/T) | 18/112 (0.14) | 131/435 (0.23) | 0.020 |
| H6PD-SPSB1, rs10746487 (T/C) | 40/88 (0.31) | 123/441 (0.22) | 0.023 |
| Intergenic, rs1985317 (T/C) | 46/84 (0.35) | 252/310 (0.45) | 0.050 |
| LINC00877, rs6771316 (A/G) | 1/129 (0.01) | 28/542 (0.05) | 0.032 |
| Variant | Chr | Variant Type (Gene) | Alleles | p-Value | |
|---|---|---|---|---|---|
| Signals at a threshold of p < 5 × 10−8 (n = 7) | All TEEs | Stroke only | |||
| rs139341092 | 20 | Intergenic | CTCA/- | 1.50 × 10−8 | |
| rs35390334 | 11 | Intronic | C > T | 2.40 × 10−8 | 3.21 × 10−9 |
| rs331532 | 11 | Intronic | G > A | 2.80 × 10−8 | 2.63 × 10−9 |
| rs317777 | 11 | Exonic (OLFM5P) | G > A | 2.29 × 10−8 | 3.52 × 10−11 |
| rs147062602 | 11 | Exonic (OR51B5) | CCAGGTCTGTGGCAGCC/- | 2.47 × 10−8 | 8.70 × 10−11 |
| rs372091 | 11 | Intronic (OR51B5) | G > A | 2.95 × 10−8 | 4.43 × 10−11 |
| rs76076035 | 9 | Intronic | T > C | 3.23 × 10−8 | |
| Signals located on known genes with an association threshold of p < 5 × 10−6 (n = 34) | |||||
| rs4910823 | 11 | Intronic (TRIM5, TRIM6-TRIM34) | A > G | 7.14 × 10−8 | 3.71 × 10−10 |
| rs11785309 | 8 | Intronic (CSMD1) | T > C | 1.18 × 10−7 | 2.67 × 10−11 |
| rs73395847 | 11 | Splice acceptor (C11orf40) | C > T | 1.25 × 10−7 | 7.08 × 10−10 |
| rs12412726 | 10 | Intronic (LINC01435) | G > A | 2.57 × 10−7 | |
| rs6554634 | 5 | Transcription factor binding site (SLC6A19) | G > A | 2.64 × 10−7 | |
| rs80034548 | 11 | Exonic (OR51A1P) | G > C | 3.08 × 10−7 | 1.95 × 10−9 |
| rs1368823 | 11 | Intronic (MMP26) | C > T | 5.15 × 10−7 | 9.32 × 10−9 |
| rs1455957 | 11 | Linked with rs2472530 in OR52A5 | A > G | 5.37 × 10−7 | 4.70 × 10−9 |
| rs73388885 | 11 | 3 prime UTR variant (OR51E2) | G > A | 5.46 × 10−7 | 1.05 × 10−9 |
| rs7933549 | 11 | Missense (OR51V1) | G > A | 1.08 × 10−6 | 3.17 × 10−8 |
| rs73405065 | 11 | Intronic (MMP26) | G > T | 1.10 × 10−6 | 5.57 × 10−9 |
| rs62071691 | 11 | Intronic (CAMKK1) | A > G | 1.10 × 10−6 | |
| rs9667878 | 11 | Upstream variant (OR51V1) | C > T | 1.11 × 10−6 | 3.25 × 10−8 |
| rs73402629 | 11 | Upstream transcript enhancer (HBG1) | C > A | 1.41 × 10−6 | |
| rs11035718 | 11 | Linked with rs2472530 in OR52A5 | G > A | 1.63 × 10−6 | 2.33 × 10−8 |
| rs4525262 | 11 | Intronic (TRIM22) | A > C | 1.94 × 10−6 | 1.41 × 10−10 |
| rs73405021 | 11 | Intronic (MMP26) | A > G | 2.06 × 10−6 | 2.24 × 10−8 |
| rs317781 | 11 | Intronic (UBQLN3) | A > C | 2.06 × 10−6 | 9.84 × 10−10 |
| rs35504021 | 9 | Intronic (DOCK8) | G > A | 2.36 × 10−6 | |
| rs1566860 | 8 | Intronic (CSMD1) | G > A | 2.53 × 10−6 | 3.49 × 10−8 |
| rs141682404 | 6 | Intronic (PACRG) | C > T | 2.74 × 10−6 | |
| rs73402631 | 11 | Upstream variant (HBG1) | C > G | 2.98 × 10−6 | |
| rs10742697 | 11 | Upstream variant (UBQLN3) | C > A | 3.16 × 10−6 | 1.63 × 10−9 |
| rs2213170 | 11 | Intronic (HBE1) | A > G | 3.16 × 10−6 | |
| rs2071348 | 11 | Enhancer (HBBP1) | T > G | 3.25 × 10−6 | 4.19 × 10−9 |
| rs2599587 | 11 | Intronic (TRIM21) | C > A | 3.68 × 10−6 | |
| rs73427713 | 11 | Downstream variant (OR52K1) | A > G | 3.80 × 10−6 | 4.43 × 10−8 |
| rs7026337 | 9 | Intronic (NTRK2) | A > G | 3.93 × 10−6 | |
| rs116926964 | 11 | Intronic (ATG16L2) | G > T | 3.93 × 10−6 | |
| rs36030093 | 8 | Intronic (LOC105375776) | C > T | 4.21 × 10−6 | |
| rs11033350 | 11 | Intronic (OR51E2) | A > G | 4.34 × 10−6 | 2.77 × 10−8 |
| rs12286769 | 11 | Upstream variant (OR52K2) | C > T | 4.62 × 10−6 | 2.33 × 10−8 |
| rs72981995 | 6 | Intronic (LOC105378031) | A > C | 4.85 × 10−6 | |
| rs967664 | 11 | Downstream variant (OR52T1P) | T > G | 4.92 × 10−6 | 1.48 × 10−8 |
| Number of Observations | Haplotype | Haplotype Code | Inherited Cases (%) | Inherited Controls (%) | p-Value |
|---|---|---|---|---|---|
| 11 | CC-GG-ins-AG-del-TT-CC-TT-GG-GG | haplo-12 | 0 (0.0) | 11 (16.92) | 0.14 |
| 20 | CC-GG-ins-GG-del-CT-CC-TT-GG-GG | haplo-17 | 2 (3.08) | 18 (27.69) | 0.39 |
| 8 | CC-GG-ins-GG-del-TT-CC-TC-GG-GG | haplo-20 | 1 (1.54) | 7 (10.77) | 1.0 |
| 63 | CC-GG-ins-GG-del-TT-CC-TT-GG-GG | haplo-22 | 5 (7.69) | 58 (89.23) | 0.019 |
| 5 | CC-GG-ins-GG-ins-TT-CC-TT-GG-GG | haplo-25 | 1 (1.54) | 4 (6.15) | 1.0 |
| 5 | TC-AA-del-AA-del-TT-TC-CC-CC-AA | haplo-40 | 0 (0.0) | 5 (7.69) | 0.59 |
| 5 | TC-AG-W-AA-del-TT-TC-CC-CC-AA | haplo-50 | 0 (0.0) | 5 (7.69) | 0.59 |
| 5 | TC-GG-ins-GG-del-TT-CC-TC-GG-GG | haplo-77 | 0 (0.0) | 5 (7.69) | 0.59 |
| 5 | TC-GG-ins-GG-del-TT-CC-TT-GG-GG | haplo-78 | 0 (0.0) | 5 (7.69) | 0.59 |
| 15 | TT-AA-del-AA-del-CT-TT-CC-CC-AA | haplo-84 | 5 (7.69) | 10 (15.38) | 0.17 |
| 23 | TT-AA-del-AA-del-TT-TT-CC-CC-AA | haplo-89 | 8 (12.31) | 15 (23.08) | 0.051 |
| 7 | TT-AA-del-AA-ins-CT-TT-CC-CC-AA | haplo-91 | 4 (6.15) | 3 (4.62) | 0.024 |
| 9 | TT-AG-W-AA-del-TT-TT-CC-CC-AA | haplo-102 | 0 (0.0) | 9 (13.85) | 0.21 |
| 5 | TT-AG-W-AG-del-TT-TT-CC-CC-AA | haplo-108 | 0 (0.0) | 5 (7.69) | 0.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshabeeb, M.A.; Alwadaani, D.; Al Qahtani, F.H.; Abohelaika, S.; Alzahrani, M.; Al Zayed, A.; Al Saeed, H.H.; Al Ajmi, H.; Alsomaie, B.; Rashid, M.; et al. Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease. Genes 2023, 14, 1919. https://doi.org/10.3390/genes14101919
Alshabeeb MA, Alwadaani D, Al Qahtani FH, Abohelaika S, Alzahrani M, Al Zayed A, Al Saeed HH, Al Ajmi H, Alsomaie B, Rashid M, et al. Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease. Genes. 2023; 14(10):1919. https://doi.org/10.3390/genes14101919
Chicago/Turabian StyleAlshabeeb, Mohammad A., Deemah Alwadaani, Farjah H. Al Qahtani, Salah Abohelaika, Mohsen Alzahrani, Abdullah Al Zayed, Hussain H. Al Saeed, Hala Al Ajmi, Barrak Alsomaie, Mamoon Rashid, and et al. 2023. "Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease" Genes 14, no. 10: 1919. https://doi.org/10.3390/genes14101919
APA StyleAlshabeeb, M. A., Alwadaani, D., Al Qahtani, F. H., Abohelaika, S., Alzahrani, M., Al Zayed, A., Al Saeed, H. H., Al Ajmi, H., Alsomaie, B., Rashid, M., & Daly, A. K. (2023). Impact of Genetic Variations on Thromboembolic Risk in Saudis with Sickle Cell Disease. Genes, 14(10), 1919. https://doi.org/10.3390/genes14101919