1. Introduction
The accuracy and completeness of reference genomes have an important influence on the accuracy of clinical next-generation sequencing (NGS) data analyses. Since the first human reference genome was published in 2001 by the Genome Reference Consortium (GRC), several updated versions of the human reference genome have been released with subsequent incremental improvement (
https://www.ncbi.nlm.nih.gov/grc, accessed on 10 August 2023). The most recent build is the Genome Reference Consortium Human Build 38 (GRCh38, released in 2013), which is also referred to as Human Genome 38 (hg38). GRCh38 was released with improvements over GRCh37 (also known as hg19, released in 2009) in terms of accuracy and completeness. Previous studies reported that, compared with GRCh37, GRCh38 updated 8000 nucleotides; filled in numerous gaps; added sequences of centromeres, telomeres, and the mitochondrial genome; and encompassed population-specific genomic contents [
1,
2].
The different builds of the reference genome not only result in different genome assemblies but also impact genomic analyses and variant classification. Also, the reference allele might not represent the major allele, according to builds, because the reference genome is determined by a very small group of individuals. For instance, the Genome Aggregation Database reports that the allele frequency of the factor V Leiden (FVL) variant [c.1601G > A (p.Arg534Gln) in the
F5 gene] in GRCh37 (Chr1: 169549811) and GRCh38 (Chr1: 169519049) was 98.1% and 1.8%, respectively (
https://gnomad.broadinstitute.org/variant/1-169549811-C-T?dataset=gnomad_r3, accessed on 10 August 2023) [
3]. The FVL variant may be erroneously defined as the reference allele based on GRCh37. This means that the pathogenic risk alleles based on GRCh38 could be defined as non-pathogenic common alleles based on GRCh37. Furthermore, it has been reported that the use of GRCh37 could result in the false detection of variants in clinically significant genes such as
KCNE1 (Jervell-Lange-Nielsen syndrome 2, long-QT syndrome 5),
NOTCH2 (Alagille syndrome 2; Hajdu-Cheney syndrome), and
SIK1 (developmental and epileptic encephalopathy 30) [
2,
4]. In addition, sequence differences between GRCh37 and GRCh38 have been reported to be found in disease-related genes such as
NCF1 (Chronic granulomatous disease 1, autosomal recessive),
ADAMTSL2 (Geleophysic dysplasia 1), and
RPS17 (Diamond–Blackfan anemia 4) [
2]. Therefore, if GRCh37 continues to be used, there are risks of missing or inaccurately interpreting clinically significant variations. Several studies regarding GRCh38 alignment have reported that the implementation of GRCh38 could produce more accurate and consistent genomic data through the analysis of single nucleotide variations (SNVs), insertions/deletions/duplications (indels/dup), structural variants, and copy number variations, compared with those based on GRCh37 [
4,
5].
To date, GRCh38 has not been widely adopted and GRCh37 is still extensively used in sequencing data analyses. There are clear benefits and disadvantages of using GRCh38. When switching reference genomes from GRCh37 to GRCh38, the accuracy of NGS data analyses is expected to improve with the improvement of the accuracy and completeness of the reference genome. However, there is a disadvantage in that it is necessary to revalidate the bioinformatic pipeline for the application of GRCh38. This requires additional investment of time, cost, human resources, and computational resources to facilitate the migration. In addition, the raw sequence data for re-alignment can be large, and they are not always accessible. A recent survey demonstrated that clinical laboratories hesitate to migrate to GRCh38; only 7% (2/28) of laboratories migrated to GRCh38 [
6]. Furthermore, more than half of the laboratories aligning with GRCh37 (58%, 15/28) responded that they had no plan to change to GRCh38 [
6] because their routine bioinformatics pipelines are based on GRCh37, and migration to GRCh38 requires revalidation of the total bioinformatics pipelines [
6]. However, if clinical laboratories continuously stick to GRCh37, there would be the risk of missing clinically significant variants as well as the false detection of pathogenic variants [
3,
4,
7,
8]. Currently, there is a need for a fast and convenient approach to implement GRCh38.
There are two approaches for reference genome conversion from GRCh37 to GRCh38. One is a time-consuming and computationally expensive re-alignment procedure, and the other is the simple use of liftover tools as an alternative approach to re-alignment [
1,
9,
10,
11]. Although re-alignment provides the most accurate results, it may not be practical in clinical laboratories. Several liftover tools such as CrossMap (
http://asia.ensembl.org/Homo_sapiens/Tools/AssemblyConverter?db=core, accessed on 10 August 2023), the National Center for Biotechnology Information (NCBI) Remap (
https://www.ncbi.nlm.nih.gov/genome/tools/remap, accessed on 10 August 2023), and the University of California Santa Cruz (UCSC) liftOver (
https://genome.ucsc.edu/cgi-bin/hgLiftOver, accessed on 10 August 2023), rtracklayer::liftover (
https://www.bioconductor.org/help/workflows/liftOver/, accessed on 10 August 2023), flo (
https://github.com/wurmlab/flo, accessed on 10 August 2023), and segment_liftover (
https://github.com/baudisgroup/segment-liftover, accessed on 10 August 2023) have been developed to convert genome coordinates from one build to another [
9,
10,
11,
12,
13]. Compared with the alignment approach, liftover has advantages in terms of simplicity and versatility. Liftover tools are easy to use. For example, CrossMap, NCBIRemap, and UCSC liftOver were freely available on the website. In addition, CrossMap and UCSC liftOver only require storage-friendly chain files, which describe pairwise alignment between genome assemblies. Most of the liftover tools also support the most commonly used file types including Browser Extensible Data (BED), General Feature Format (GFF)/Gene Transfer Format (GTF), VCF, Binary Alignment Map (BAM)/Sequence Alignment MAP (SAM), BigWig [
9,
10,
11,
12,
13]. The liftover tools can be used in any situation where any reference genome conversion is required, including cross-species mapping [
9,
10,
11,
12,
13].
Different liftover tools can be classified into two categories according to a strategy to preserve the segment: integrity preserved approach (e.g., NCBI Remap) vs. non-integrity preserved approach (e.g., CrossMap, UCSC liftOver, rtracklayer::liftover, flo, and segment_liftover) [
9,
10,
11,
12,
13]. Currently, the accuracy and limitations of liftover tools are not well known [
9,
10,
11,
12,
13]. Furthermore, the performance of liftover tools using a large set of clinical variants has not yet been extensively explored [
4,
13,
14,
15]. This study aimed to investigate the accuracy of three liftover tools, CrossMap, NCBI Remap, and UCSC liftOver for conversion to GRCh38 from GRCh37, comparing the re-alignment approach.
4. Discussion
We have evaluated three liftover tools for genome conversion using clinical variants from ClinVar. The ClinVar is one of the most commonly used clinical databases for variant curation and interpretation. Currently, studies regarding genome conversion for clinical variants using liftover tools have not been rigorously investigated. Pan et al., have reported the conversion rate (average 99%) of SNV from NA12878 from GRCh37 to GRCh38 using LiftoverVcf from the Picard package and CrossMap [
14]. Ormond et al. have investigated the conversion failure rates of 0.14% from GRCh37 to GRCh38 [
15]. These two studies have been performed using reference materials such as NA12878, and they do not represent clinical variants [
14,
15]. In addition, previous studies focused on the conversion of SNVs and did not consider indels/dup [
14,
15]. To date, there has been only one study regarding the genome conversion of a limited set of clinical variants (n = 158) using UCSC liftOver [
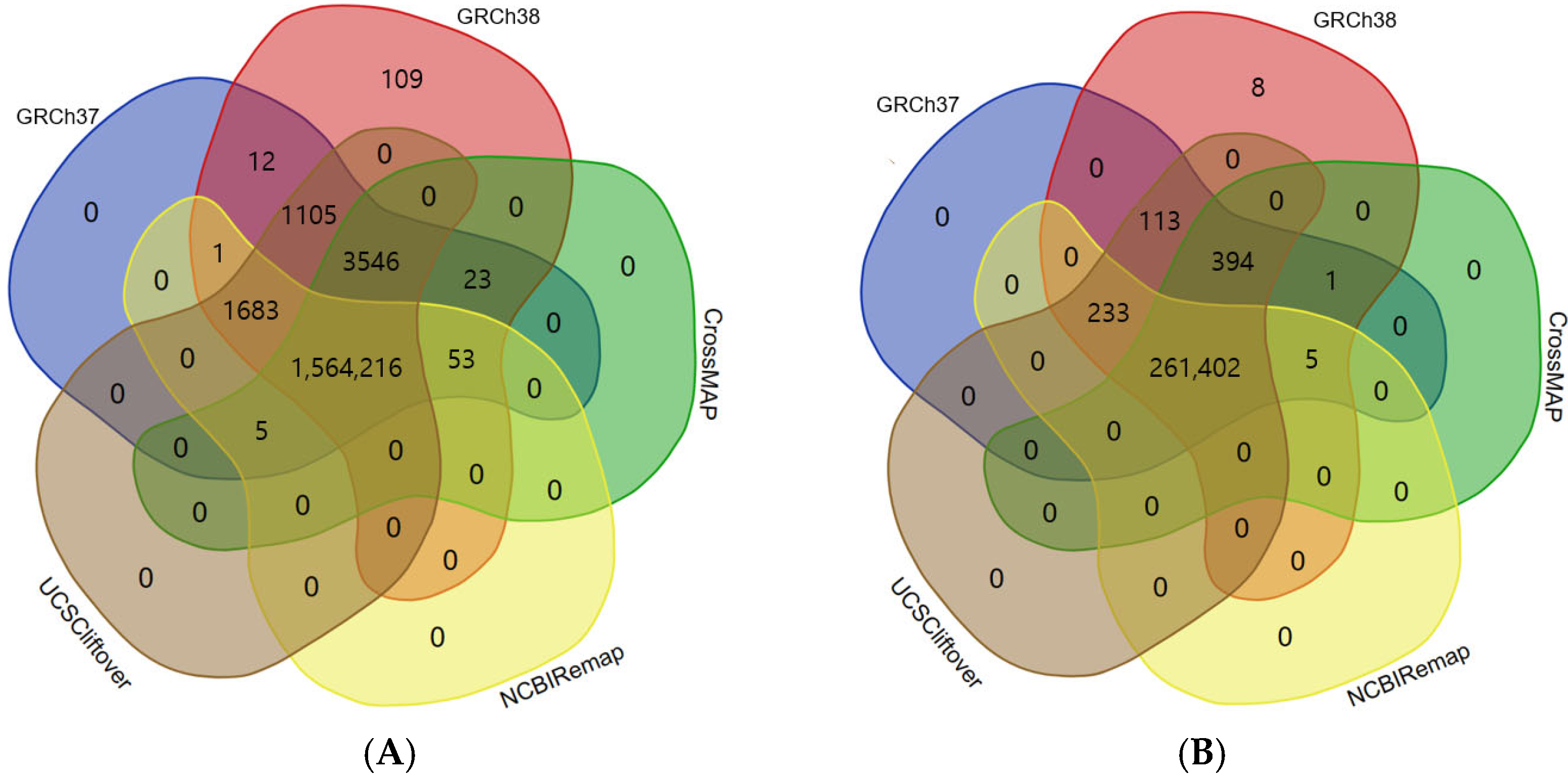
4]. We investigated the conversion rate and accuracy of liftover tools using a large set of clinical variants from ClinVar (GRCh37-aligned ClinVar variants and GRCh38-aligned ClinVar variants) including indels/dup as well as SNVs.
There were significant discrepancies between GRCh37-aligned ClinVar variants and GRCh38-aligned ClinVar variants. We found that indels/dup were added more in the GRCh38-aligned VCF file [(indels/dup (n = 72) vs. SNVs (n = 37))]. That means that using GRCh38 can produce more indels/dup compared to GRCh37. A number of variants newly included in the GRCh38-aligned VCF file were in clinically significant genes, including
RBM8A (Thrombocytopenia-absent radius syndrome),
CHD1L (HCCs and other solid tumors),
NUF2 (4p partial monosomy syndrome),
ALMS1 (Alstrom syndrome),
TTN (Tibial muscular dystrophy, Myopathy, myofibrillar, 9, with early respiratory failure, Autosomal recessive limb-girdle muscular dystrophy type 2J, Early onset myopathy with fatal cardiomyopathy),
TFAP2B (Chronic intestinal pseudo-obstruction),
LAMA2 (Merosin deficient congenital muscular dystrophy),
FKBP6 (Male infertility, Spermatogenic failure 77),
MUC3A (Lung cancer),
CFTR (Cystic fibrosis, CFTR-related disorders, Obstructive azoospermia, Hereditary pancreatitis, Congenital bilateral aplasia of vas deferens from CFTR mutation, Bronchiectasis with or without elevated sweat chloride 1, modifier of),
NEFL (Charcot-Marie-Tooth disease type 2E),
OPLAH (5-Oxoprolinase deficiency),
RECQL4 (Baller-Gerold syndrome),
PTCH1 (Gorlin syndrome),
MUC2 (Small cell lung carcinoma),
PAX6 (Aniridia 1),
OR8J1 (Premature ovarian failure),
MEN1 (Hereditary cancer-predisposing syndrome),
SHANK2 (Autism spectrum disorder),
ATM (Ataxia-telangiectasia, Lymphoma),
C1R (Ehlers-Danlos syndrome, periodontal type 2),
LRP6 (Ductal breast carcinoma),
SLCO1B1 (Hyperbilirubinemia, Rotor type, digenic),
ALG10 (Delayed puberty),
BRCA2 (Hereditary breast ovarian cancer syndrome),
PALB2 (Hereditary cancer-predisposing syndrome),
KCNJ18 (Thyrotoxic periodic paralysis, susceptibility to, 2),
MAP3K14 (NIK deficiency),
PECAM1 (Three Vessel Coronary Disease),
DSC2 (Arrhythmogenic right ventricular dysplasia 11),
ADAMTSL5 (Ductal breast carcinoma),
LDLR (Hypercholesterolemia, familial, 1),
CEBPA (Acute myeloid leukemia),
KLK4 (Amelogenesis imperfecta),
ADAM33 (Ductal breast carcinoma),
PRNP (Inherited Creutzfeldt-Jakob disease, Gerstmann–Straussler–Scheinker syndrome, Huntington disease-like 1),
LOC102724428 (Childhood epilepsy with centrotemporal spikes),
PCNT (Microcephalic osteodysplastic primordial dwarfism type II),
MID1 (Opitz GBBB syndrome),
WDR45 (Neurodegeneration with brain iron accumulation 5),
FGF16 (Metacarpal 4-5 fusion),
GRIA3 (Syndromic X-linked intellectual disability 94),
MAMLD1 (Disorder of sexual differentiation),
L1CAM (X-linked hydrocephalus syndrome),
MECP2 (Encephalopathy, neonatal severe, Rett syndrome),
RAB39B (Early onset parkinsonism-intellectual disability syndrome), and
ABO (ABO blood group system). When limited to “authentic” ClinVar variants, we showed that only eight variants, which were newly added in the GRCh38-aligned VCF file and were not in GRCh37-aligned VCF, failed conversion by all three liftover tools. The presence of these variants may attribute false negative results when using liftover tools. Because we implemented the liftover tools using GRCh37-aligned ClinVar variants, it is reasonable that these variants were not detected as converted variants by liftover tools. To minimize false negatives, differences between reference genome builds should be considered ahead of the performance of liftover tools, because these variants are likely to be in gaps between the reference genome builds. Compared to GRCh37, GRCh38 updated 8000 nucleotides, corrected several misassembled regions, and filled in numerous gaps, indicating an ability to detect clinically significant variants with higher sensitivity [
1,
2].
To the best of our knowledge, there are no studies regarding the comparison of three liftover tools using clinical variants. One recent study has reported a high degree of correlation between liftover tools including CrossMap, NCBI Remap, and UCSC liftOver using epigenetic data such as DNA methylation and Chromatin ImmunoPrecipitation Sequencing data [
13]. In this study, we showed the similar accuracy of liftover tools using genetic data; three liftover tools could successfully convert reference genomes from GRCh37 to GRCh38 in more than 99% of ClinVar variants [CrossMap (99.82%), NCBI Remap (99.70%), and UCSC liftOver (99.99%)].
Here, we provided a list of non-converted P/LP variants identified in clinically significant genes such as
DGAT1 (Congenital diarrhea 7 with exudative enteropathy),
GPR179 (Congenital stationary night blindness),
HNF1B (Maturity onset diabetes mellitus in young, Renal cysts and diabetes syndrome),
LDLR (familial hypercholesterolemia),
MBOAT7 (Intellectual disability, autosomal recessive),
PCGF2 (Abnormality of the outer ear, Intellectual disability, Global developmental delay, Turnpenny-fry syndrome),
PQBP1 (Renpenning syndrome, Microcephaly, Intellectual disability, Delayed speech and language development, Hyperactivity),
PRPF31 (Retinal dystrophy, Retinitis pigmentosa),
PUF60 (8q24.3 microdeletion syndrome, CHARGE association),
RBP3 (Retinal dystrophy, Retinitis pigmentosa),
SLC35A2 (non-lesional focal epilepsy, SLC35A2-congenital disorder of glycosylation),
SLC39A4 (Hereditary acrodermatitis enteropathica),
SLC52A2 (Brown-Vialetto-van Laere syndrome 2),
SURF1 (Cerebellar ataxia, Dysarthria, Muscle weakness, Abnormal pyramidal sign, Cytochrome-c oxidase deficiency disease, Charcot-Marie-Tooth disease type 4K, Leigh syndrome),
TFE3 (Neurodevelopmental abnormality),
WDR45 (Delayed speech and language development, development, Neurodegeneration with brain iron accumulation 5, X-linked cerebral-cerebellar-coloboma syndrome),
ZNHIT3 (PEHO syndrome) (
Supplementary Tables S1–S3). There is a higher risk of missing clinically significant variants of these genes in clinical laboratories that remain with the use of GRCh37 as a reference genome.
However, most of the P/LP variants, except one, were converted successfully by the combined use of multiple liftover tools. One P/LP variant of NM_000527.5(LDLR):c.314-446_1187-386dup, which failed conversion by all three liftover tools, was an 8.1kb duplication variant. Considering the size of the variant, the direct re-alignment approach is more appropriate than using liftover tools for this variant; the larger the genomic segments and intervals of the variant, the lower the accuracy result of the liftover tools for genome conversion of the variant.
Here, we demonstrated that most of the non-converted variants by a single liftover tool were successfully converted by the other tools. For example, the variant of NM_002900.3(RBP3):c.1682_1686dup (p.Thr563fs) was not converted by CrossMap, whereas it was converted successfully by NCBI Remap and UCSC liftOver. Another example of NM_006331.8(EMG1):c.126dup (p.Leu43fs) was not converted by UCSC liftOver, whereas it was converted successfully by NCBI Remap and CrossMap. This suggested that the combined use of multiple liftover tools might increase the conversion rate and accuracy of the liftover tools.
There are pros and cons of using multiple liftover tools simultaneously. As shown in this study, the simultaneous use of multiple liftover tools could result in a more successful genome conversion and they could be used complementarily with each other. Fortunately, UCSC liftOver, NCBI Remap, and CrossMap are freely available on the public web. Therefore, these tools can be easily accessed, even in laboratories without bioinformatics resources. However, most liftover tools also require chain files or alignment formats and other liftover tools are relatively time consuming and computationally intensive, although they are a relatively convenient and more cost-effective approach than re-alignment [
12,
13]. Currently, the accuracy and limitations of these liftover tools have not been extensively studied. Therefore, further clinical studies should be performed to investigate the clinical utility as well as analytical utility of several liftover tools in routine clinical settings prior to the simultaneous use of liftover tools.
We investigated the type of non-converted variants. We found that the proportion of indels/dup among the total studied variants was 8.93% (23,421/262,156), while the proportion of indels/dup among non-converted variants by CrossMap, NCBI Remap, and UCSC liftOver was 13.56% (48/354), 13.95% (72/516), and 71.43% (10/14), respectively. Indels/dup are more complicated in variant size and sequence context and are likely to be more error-prone than SNVs. In addition, indels/dup frequently occur in repetitive sequences, and this can make analyses difficult. Therefore, when using the liftover tools, especially UCSC liftover, it may be helpful to consider the type of variants.
Next, we performed variant annotation and investigated whether there are highly homologous sequences such as pseudogenes and segmental duplications in the genomic regions where the variants are located. Considering that liftover tools “lift” the genome position in one reference genome build ‘over’ to another build, highly homologous sequences might also result in genome conversion failure. However, in this study, we found no pseudogene-related factors in any non-converted variants among authentic ClinVar variants.
Previous studies have investigated the problems associated with genome conversion using liftover tools [
13,
14,
15]. Pan et al., have shown that discordant SNVs had lower read depth and a higher prevalence of GC contents [
14]. Ormond et al., have reported that conversion-unstable positions were associated with gaps in the builds, contig differences between builds, and segmental duplications [
15]. Therefore, it is important to pre-exclude the problematic regions such as gapped regions before the implementation of liftover tools. Here, we pre-excluded the variants on alternative contigs to exclude conversion failure due to contig differences between builds. In the present study, we did not investigate comprehensively the genomic regions associated with conversion failure by liftover tools because we analyzed only VCF files downloaded from ClinVar. Neither FASTQ or BAM files were available in this study. We demonstrated that gaps in the reference builds or variant types such as indels/dup were associated with conversion failure. Further clinical studies to investigate the genomic characteristics associated with conversion failure would be recommended prior to the clinical use of liftover tools.
Previous studies reported that converted variants using liftover tools are not always concordant with the variants obtained from the aligned data [
13,
14,
15]. These variants might be false positive results of liftover tools because these variants were not in the VCF file obtained from alignments to GRCh38, which is considered as ground truth. In this study, five variants that were included in the GRCh37-aligned VCF file and removed from the GRCh38-aligned VCF file [for example, NM_005235.2(
ERBB4):c.83-200864_83-199104del] were also successfully converted by all three liftover tools. Despite successful conversion, these variants should not be reported if GRCh38 was chosen as a reference genome. Here, there was no discordant variants among the authentic ClinVar variants. Previous studies reported that discordant variants were noted to be in regions with segmental duplications and repetitive sequences [
13,
14,
15]. Furthermore, it has been reported that there were changes in the chromosome and genomic position of converted variants by use of liftover tools [
10,
14,
15]. Inconsistent chromosome numbers between reference genomes by different liftover tools can negatively impact downstream analyses in terms of nomenclature and classification of variants. Another explanation for discordancy is that the position in the GRCh37 is not in the GRCh38 or vice versa [
13,
14,
15]. This is because all positions are not completely comparable between the reference genomes.


{kind=link}
{kind=link}
{kind=link}