1. Introduction
Maize (
Zeal mays L.) is a significant cereal crop. Maize production around the world is adversely impacted by drought stress. The breeding of drought-tolerant genotypes is a practical way of addresing the water shortage problem. This calls for a thorough understanding of the sub-atomic elements involved in the reactions caused by drought pressure in maize, as well as genomic means such as genomic determination, hereditary designing, and genomic alteration for drought tolerant improvement. Plants’ response to stress can be categorized into four stages: alert stage, aggregation stage, support stage, and fatigue stage. The sub-atomic reactions to the drought pressure of plants include more administrative and useful assets [
1].
The transcription factor (TFs) is a significant controller protein that can unequivocally direct initiation and the restraint of target qualities. Then, plants reduce water loss as a result of partially closing stomata, effectively taking up water from underground by root foundation, and regulating metabolic cycles to correspond with existing carbon reserve. With the increment of drought, osmolytes, such as proline and solvent sugars, would aggregate to keep up with the cell turgor pressure. Moreover, various antioxidants, for example, superoxide dismutase (SOD, catalyze), reduce the toxic effect activity in reactive oxygen species (ROS) [
2].
The stacking bearing connections inside the receiver wall organization, for example, caused specific revision under the activity of the cell wall’s changing proteins, and the guideline of the phenylpropanoid pathway prompted a high collection of lignins in the cell wall. Quality delivery network policies play an essential role in plant drought reactions. Various transcriptions are responsible in various crops. In two wheat genotypes, different responses to drought stress were found bases on transcription analysis. [
3].
The results showed that in wheat the auxin and abscisic acid (ABA) receptor related to ROS and the biosynthesis of the cell wall was up regulated, while ethylene receptors were down regulated. In maize, the moderate response to drought were linked to DNA and cell cycle. It was shown that qualities engaged with metabolic pathways and the biosynthesis of optional digestion showed a reaction to lack, and the statement of a few qualities connected with ABA signal transduction were prompted under drought [
4]. The plant roots detect changes in water and soil oxygen content. It was shown that exceptional drought-responsive components were associated with various parts of the root zone in maize. The qualities connected with ROS and carbon digestion, and qualities linked to layer carrier, were essentially improved in drought-responsive qualities of 0–3 mm root peak, with 3–7 mm upon root chimp, separately. Comparable outcomes were likewise acquired in another research study utilizing two deep-rooted maize lines. Cell reinforcement chemical qualities were demonstrated to assume the essential parts in drought tolerance in the root transcriptomes analysis of essential root under drought stress, conducted by RNA sequencing, and the results demonstrated the expression of genes under low water stress. A few members from the MAPKKK family enhanced drought tolerance in leaf and maize stems, recommending the significance of the MAPKKK family under drought stress [
5].
Plant drought resilience is generally assessed by variances in aggregates and physiological and bio-chemical reactions. Drought plants can keep up with constant morphological features throughout delayed times of drought pressure by maintaining water-holding capability. According to one perspective, drought-resistant lines have some development to do to increase their capacity to keep moisture content in their root system. The entire root development of maize may be made to conduct less research on the soil profile and extend into more soil by reducing the number of particular cortical cells in maize, while simultaneously improving their size. This allows maize to uptake greater water from the soil. The thickness of the leaves may be reduced, and the opening of the stomata in the plants can be regulated to increase the plant’s resistance to drought [
6].
Furthermore, during conditions of drought, plants are able to maintain the homeostasis of their cells via the process of osmoregulation, which is orchestrated by soluble sugars. This helps to decrease the amount of water that is likely to be present in the cells. Water stress also mandates the peroxide-rummaging catalyst structure to eradicate the quantity of responsive oxygen species incited by drought. This is necessary since ROS may cause damage to the cell film structures, which can ultimately lead to cell death. The activity of the cell reinforcement (nonenzymatic and enzymatic) framework addresses an important file for evaluating maize’s resistance to drought. Biochemical and physiological alterations between deep-rooted lines and transgenic lines have allowed for the identification of drought-tolerant lines. Despite this, an exclusive explanation for maize’s drought resistance could not be provided by these advances, since different types had different genetic basis [
7].
It is realistic that the drought reaction of plants includes a chain of reactions. In this approach, it is planned that reading the article would help agricultural plants become more resilient to drought by providing a realistic illustration of these features. To date a couple of droughts, and tolerant features, for example, H21 and 478 in maize, were distinguished by conventional sequencing strategies and practically depicted. Many key qualities were suggested in drought-tolerant maize; however, an overall significant stretch was expected to explain the elements of these drought reaction qualities. RNA-seq is an incredible technique for the enormous scope ID of drought-responsive attributes with minimal expense, high throughput, and high responsiveness. It can work with mining crucial drought-tolerant qualities in plants, e.g., maize [
8].
For instance, research findings have demonstrated that the overexpression of cell wall oxidative metabolism characteristics makes it possible for maize recombinant inculcated lines to procure water stress flexibility under water shortages; the attributes associated with cell wall reformation are involved with water stress chemical reactions in a drought resistant corn line; and the compositions of proteins and carbohydrates are associated with water stress support. Through the process of lighting seeds from an intrinsic maize line, we were able to create a deficit resistant line for this study. To investigate the components of drought opposition of H21, we have analyzed physiological, bio-chemical, and transcriptomic changes between H21 and 478 lines. Our outcomes would feature the drought-tolerant marks of H21 and add to the distinguishing resilient and useful investigations of novel drought resilience qualities in maize [
9].
2. Materials and Methods
2.1. Plant Material and Experimental Design
Seeds of maize inbred lines, drought-tolerant (478) and drought-sensitive (H21) were given by College of Agronomy, Gansu Agricultural University, Lanzhou, China. Seeds were sown in pots holding standard preserving soil. The pots were kept in a climate that was sustained at a temperature of 25 ± 5 degrees Celsius, with a relative humidity of 65 ± 5 percent, and a light/dark cycle of 16/8 h. All the plants were adequately sprinkled to keep the moistness level persistent till 10 days after emergence; after that, drought treatments began. Treatments included two different degrees of drought conditions (no stress and stress), as well as three different levels of BR (0, 25, and 50 mL). To simulate the effects of drought, an application of fifty milliliters of 20 percent PEG 6000 every 3 days was made, whereas the control area received applications of pure water. For the transcriptome study, plants were chosen after they had reached one of four fully formed leaf stages. Samples were then kept at −80 degrees Celsius in liquid nitrogen for 7 days. The experiment was laid out in a completely randomized design (CRD), and there were three separate replications of the experiment.
2.2. RNA Extraction, Construction of Library, and Sequencing
Pure and simple RNA was detached using Trizol reagent (Thermo Fisher, Carlsbad, CA, USA, 15596018). Total RNA quantity and quality was determined using the Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, Santa Clara, CA, USA, 5067-1511) and RNA assays from the first order with a RIN greater than 7, 0 was used in building sequencing libraries. After removal of the whole RNA, the mRNA residual in the purified RNA (5 μg) was cleaned by two cycles of filtration with Dynabeads Oligo (dT) (Thermo Fisher, Carlsbad, CA, USA). Once filtered, the mRNA was cleaved into shorter fractions using higher temperature divalent cations (using the NEB Magnesium RNA Fragmentation Module, cat. e6150, Ipswich, MA, USA) for 5–7 min at 94 °C. The cut RNA fragments were then reverse disassembled to generate cDNA using the Super ScriptTM II reverse transcriptase enzyme (Invitrogen, cat. 1896649, Carlsbad, CA, USA). The cDNA was then used to combine the second desert DNA named U with DNA polymerase I from E. coli (NEB, cat.m0209, USA) and RNase H (NEB, cat.m0297, USA) and dUTP solution (Thermo Fisher, Carlsbad, CA, USA, USA, ref. R0133). The crude ends of each thread receive an A-base coating, which prepares the fibers for tightening into the taped connectors.
Each particular connector was constructed with a T-base shade so that it could ligate to the A-followed segmented DNA and complete the connection. AM Pure XP dabs were used for the purpose of carrying out size verification after twofold document connections had been affixed to the various components. Following the power labile UD Genzyme (NEB, cat.m0280, USA) treatment of the U-named second-deserted DNAs, the ligated things were escalated with PCR using the following conditions: starting denaturation at 95 °C for 3 min; 8 examples of denaturation at 98 °C for 15 s, treating at 60 °C for 15 s, and increase at 72 °C for 30 s; and a short time later last extension at The expansion sizes of the cDNA libraries that came before this one varied, on average, from 300 to 50 base pairs in length. We finished the 2 × 150 bp matched end sequencing (PE150) utilizing an Illumina NovaseqTM 6000 after the shipper advised that we do so (LC-Bio Technology Co., Ltd., Hangzhou, China).
2.3. Sequence and Filtering of Clean Reads
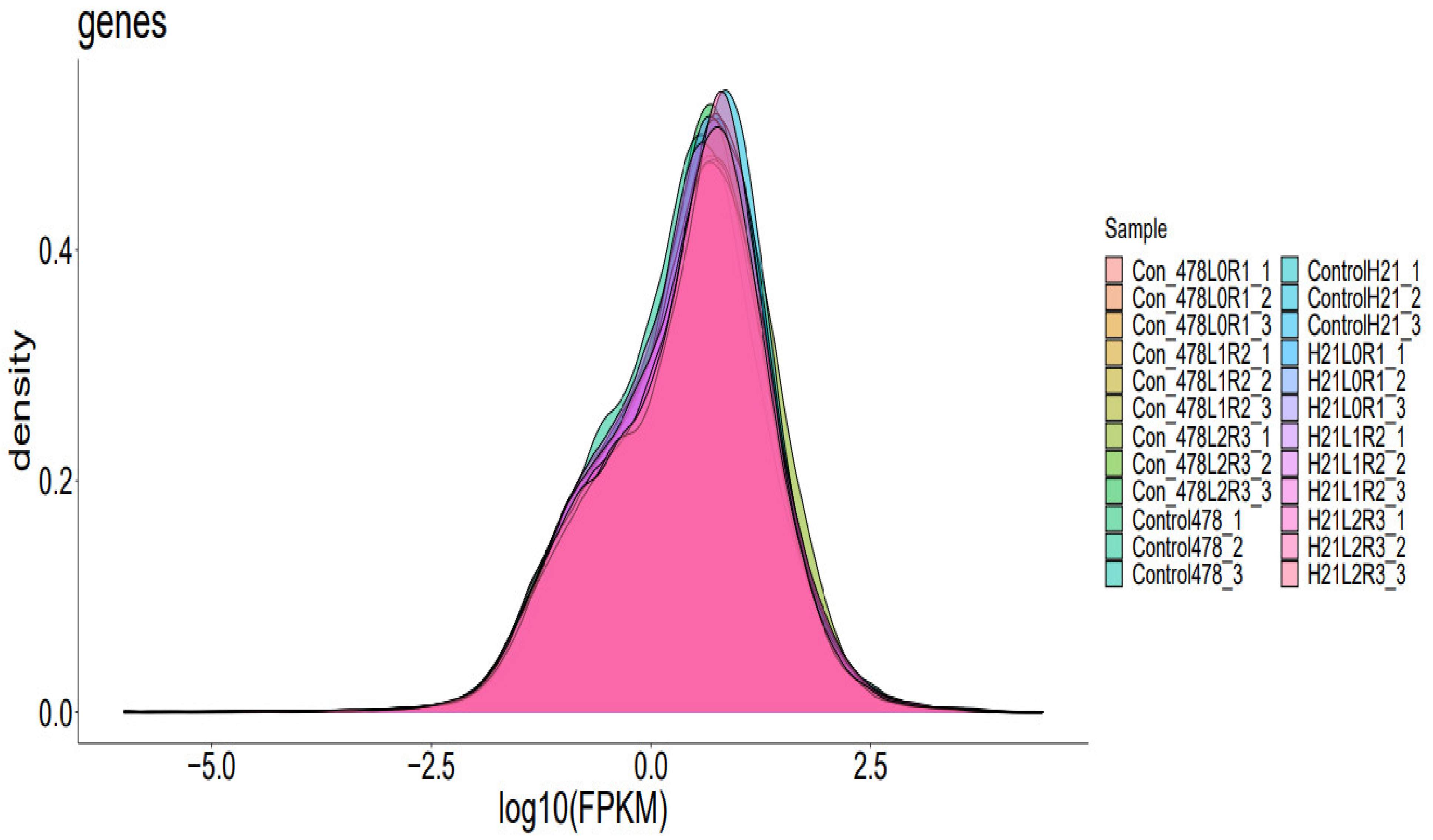
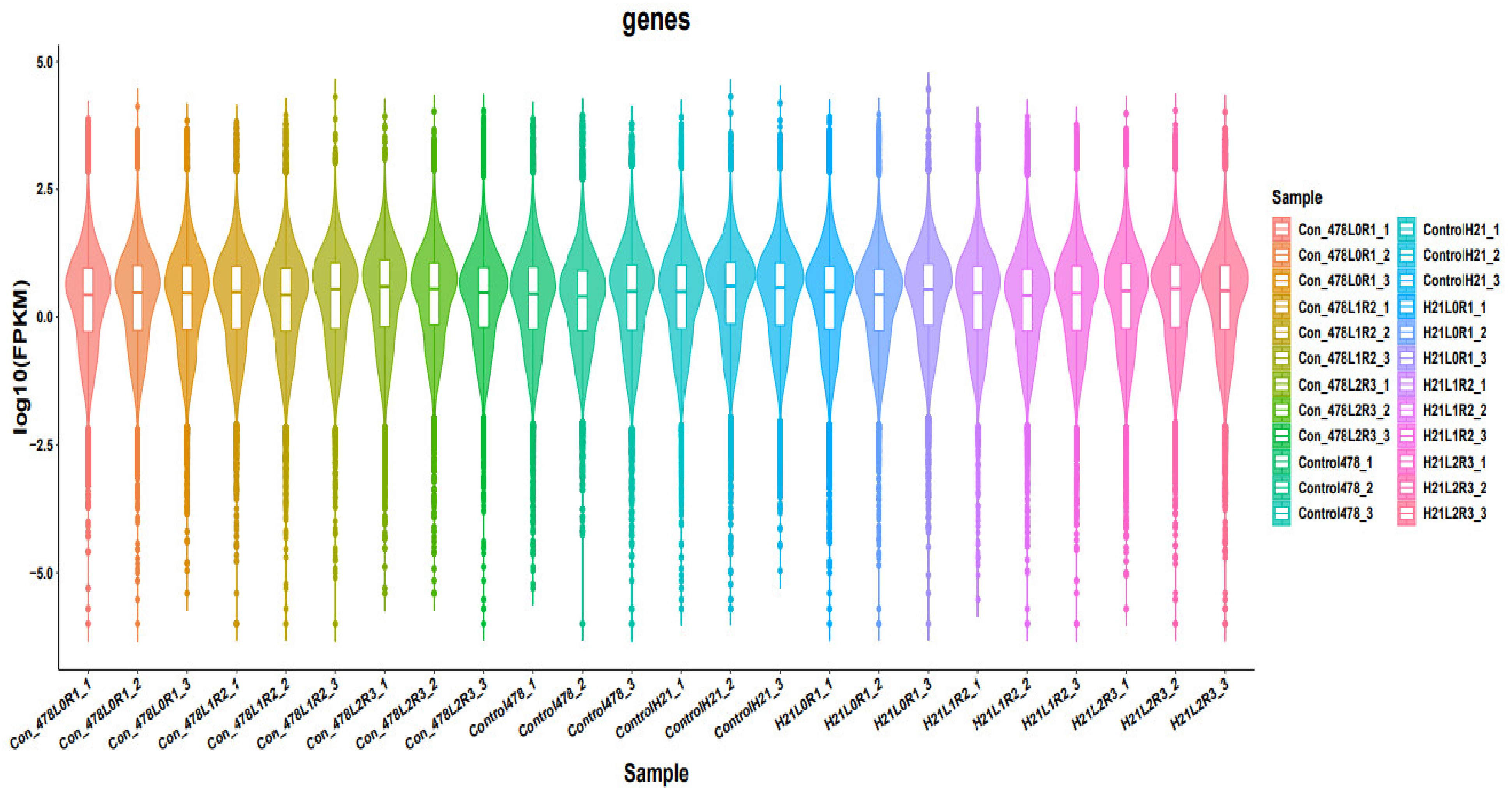
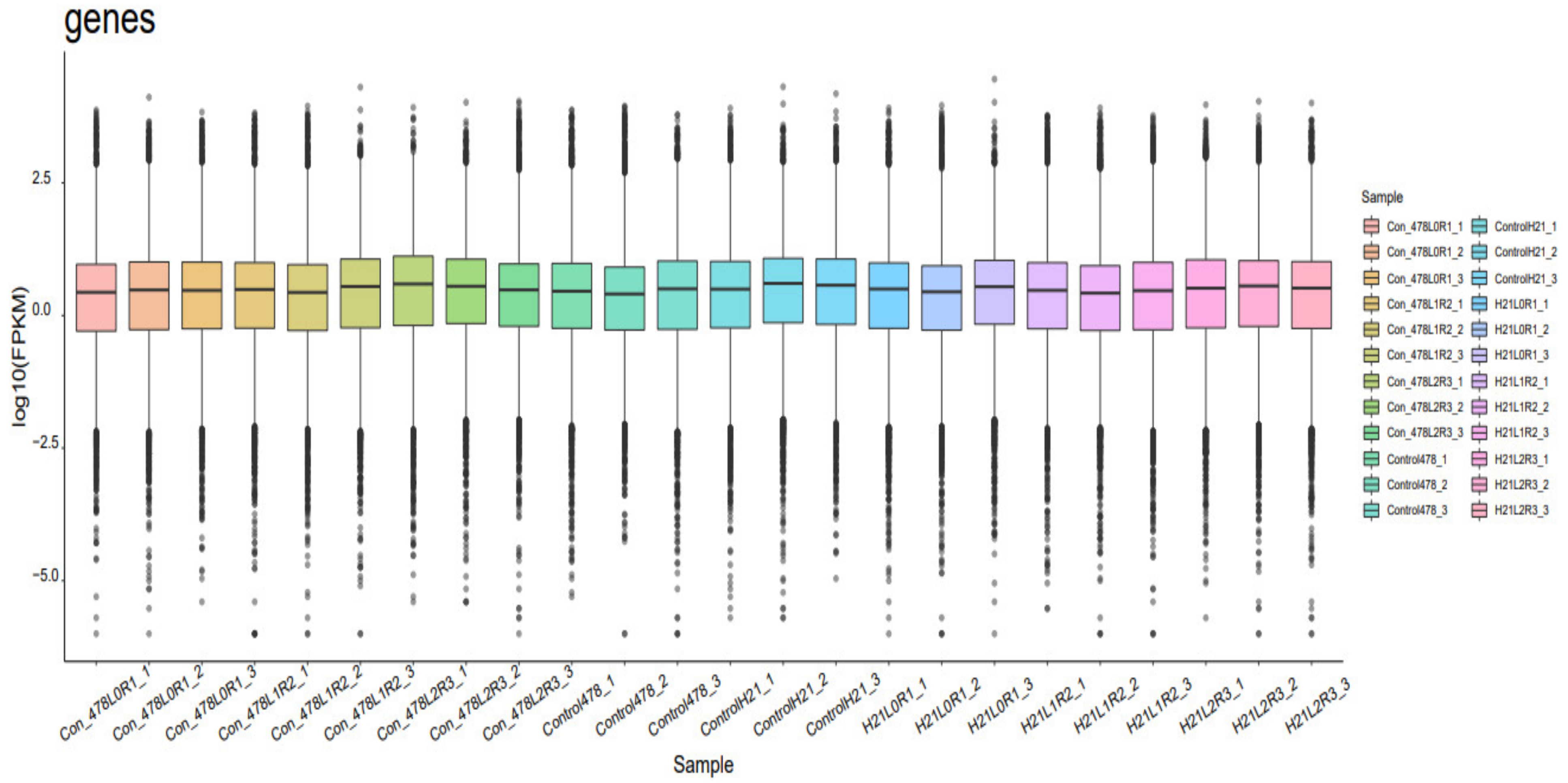
A cDNA library was created by design and sequenced utilizing pooled RNA from experiments conducted using the Illumina Novaseq TM 6000 succession stage. We used the Illumina paired-end RNA-seq method to sequence the transcriptome; readings generated from the sequencing equipment comprise raw read out with devices or low-quality bases, which have an impact on the subsequent assembly and analysis (Martin, 2011). As a result, readings were further filtered using Cutadapt to provide high-quality clean reads. The following were the criteria: eliminating reads that contain adapters, poly A and poly, more than 5% of unknown nucleotides (N), low-quality reads that contain more than 20% of low-quality (Q-value 20) bases, and reads that contain adapters. Then, FastQC was used to assess the sequence’s quality. It was noted that the data’s Q20, Q30, and GC-content had not been tainted. Following that, readings with clean endings totaling Gigabase Pairs were produced [
7]. The NCBI Gene Expression Omnibus (GEO) databases or the NCBI Short Read Archive (SRA) databases have received the raw sequencing data along with accession numbers. These two archives each have an accession number.
5. Discussion
The current study has resulted in the development of a maize freak, H21 by 478, that is resistant to drought. In comparison to its wild type 478, it showed a higher level of drought resistance when it was subjected to drought conditions. Between H21 and 478, there continued to be significant differences in the analytical files (
Figure 1). The relative water content of the leaf, often known as RWC, is a metric that is commonly used to identify the drought resistance of several agricultural crops, including corn. Under drought conditions, H21 showed a higher RWC in its leaves than 478 did; for example, it had a higher ability to maintain high moisture content. The amino acid proline and the sugar that is easily dissolved both play important roles in the osmotic guideline, which is connected with drought tolerance (
Figure 2). When exposed to drought, the leaves of the H21 variety contain much more proline and soluble carbohydrates than those of the 478 variety. When maize is subjected to drought stress during the transformation from somatic to reproductive development, the plant shows a considerable reduction in grain output. In order to create drought-resistant varieties, it is essential to have a comprehensive understanding of the biochemical and gene regulatory networks involved in corn’s tolerance to drought conditions during these various stages of development. This will allow for the development of cultivars that are more resistant to drought. Even with recent advances in molecular biology tools, the complex adaptive mechanisms that underlie water deficiency stress resistance from cellular proliferation to vegetative plant growth continue to be an enigma [
17].
A better productivity of the antioxidative safeguard framework might safeguard photosynthetic colors, proteins, and genetic material from abundant ROS loss. Feline action is conversely relative to MDA content under serious drought tolerance. The examiner of CAT action and MDA content demonstrated that H21 experienced a smaller amount of ROS harm than 478 under drought. Light dispersion in plant shade can be improved in the field by diminishing H21 content (
Figure 3). Then again, nitrogen and energy saved by a fading H21 combination would upgrade the particular reactions to drought. In this manner, low H21 content in the leaves of H21 may be a piece of its drought variation methodology [
18]. More significant, the levels of RWC, osmolyte collection, cell reinforcement exercises, photosynthetic effectiveness, and a lower degree of MDA added to the drought tolerance of H21 than 478 (
Figure 4).
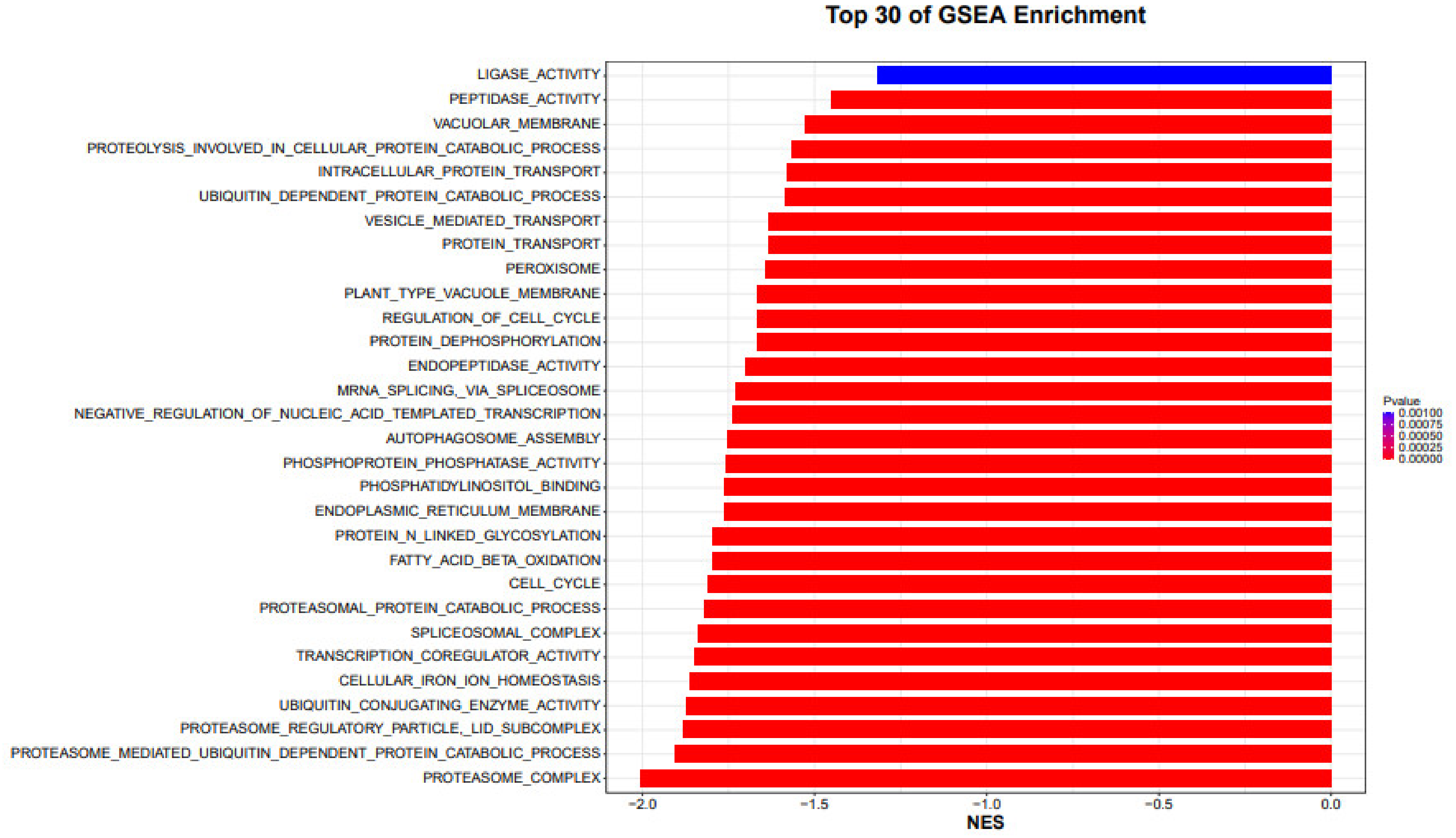
Evaluating the transcriptome state and mining genes associated to drought tolerance in maize may both be accomplished successfully with the use of genome-wide gene expression profiling, performed by RNA-Seq [
5]. Numerous genes that are sensitive to drought or that exhibit genotypically varied expression across the two lines with divergent resistance to dry conditions were found in our investigation [
11]. The opposite transcriptional regulatory predisposition that reacted to water stress in maize and the diversified gene function enhancement between drought resistant and susceptible lines providing a picture of the regulatory network in maize’s transcriptome that was linked to water stress. This was shown by the fact that there was a noteworthy variance among the two types of lines. The overrepresentation of transcriptional regulators is common in most cases, particularly sensitive genes and genotypically differently expressed genes, in the tolerant line revealed a varied mechanism for maize drought tolerance [
6].
Solvent sugars such as sucrose, galactose, maltotriose, and oligosaccharides, are the types of sugars that accept dynamic fractions in osmotic adjustment when there is a drought. In drought-tolerant wheat, the expanded delivery of dissoluble sugar blend-related qualities made sense of its drought tolerance. In the current research, the solvent sugar content in H21 was greater than that in 478 under the influence and drought disorders (
Figure 5). Obviously, H21 has an effective drought transformation system mostly founded on the osmoregulation of dissolvable sugar [
5]. In the present research, the discharge of photosynthesis-related qualities in H21 was practically unaffected, hence H21 could compose solvent sugar all the more effectively under drought conditions. In order to identify important regulatory genes and gene co-expression pathways that are involved in the maize plant’s response to environmental stress, we used RNA-seq based technique to undertake a thorough relative transcriptome study of drought-resistant lines from the somatic to propagative development stages. In order to support the RNA-seq data, we also measured photosynthetic parameters. In addition, a functional validation study using qRT-PCR provided evidence for the differential expression of the genes that were discovered. Our insights not only improve our knowledge of the processes that enable maize to survive the effects of drought stress, but they also provide a genetic resource that is both efficient and cost-effective for the genetic improvement of corn (
Figure 6).
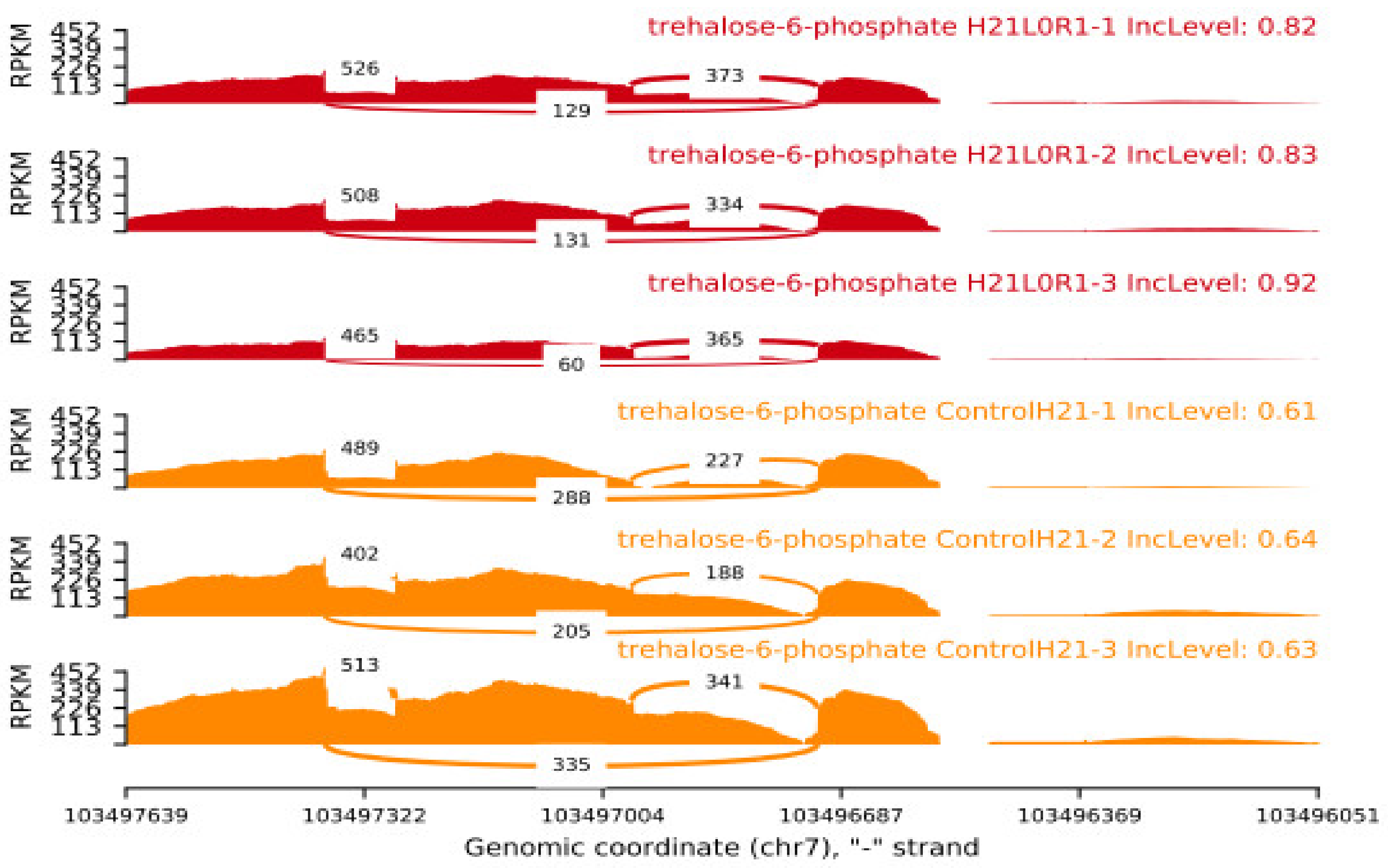
At the level of the transcriptome, H21 guided the activity of a specific component of the sugar solution. Glucose has the potential to form trehalose through a 1,1-glycosidic bond, so mitigating the damage produced by high contents of trehalose-6-phosphate under conditions of prolonged drought. In the current study, phosphatase’s behavior was restrained in 478, while their expression displayed a sharp fluctuation under drought (log 2 overlay changes from 3.46 to 7.63), indicating that the increased substance of solvent sugars in H21 probably will not be expected to the improved trehalose combination (
Figure 7). Additionally, both 478 and H21’s -amylase outputs were activated by dryness, indicating that the leaves’ starch could be broken down into maltose [
19].
Using two breeding lines of corn that had varied levels of tolerance to droughts, researchers used RNA-Seq technology to investigate an amount of DNA that were susceptible to drought, as well as proteins that showed differential expression across variants [
2]. Between the resistant and susceptible lines, researchers discovered significant differences in the regulatory patterns and operational abundance of these proteins. The fact that drought resistance and resilience were both enhanced by the overrepresentation of certain transcription factors in the tolerant line [
18] demonstrates how important these variables are. Our findings subsidize to an improved considerate of the molecular processes involved in drought response and to the identification of candidate genes for future research in the quest to improve maize’s resistance to drought [
14].
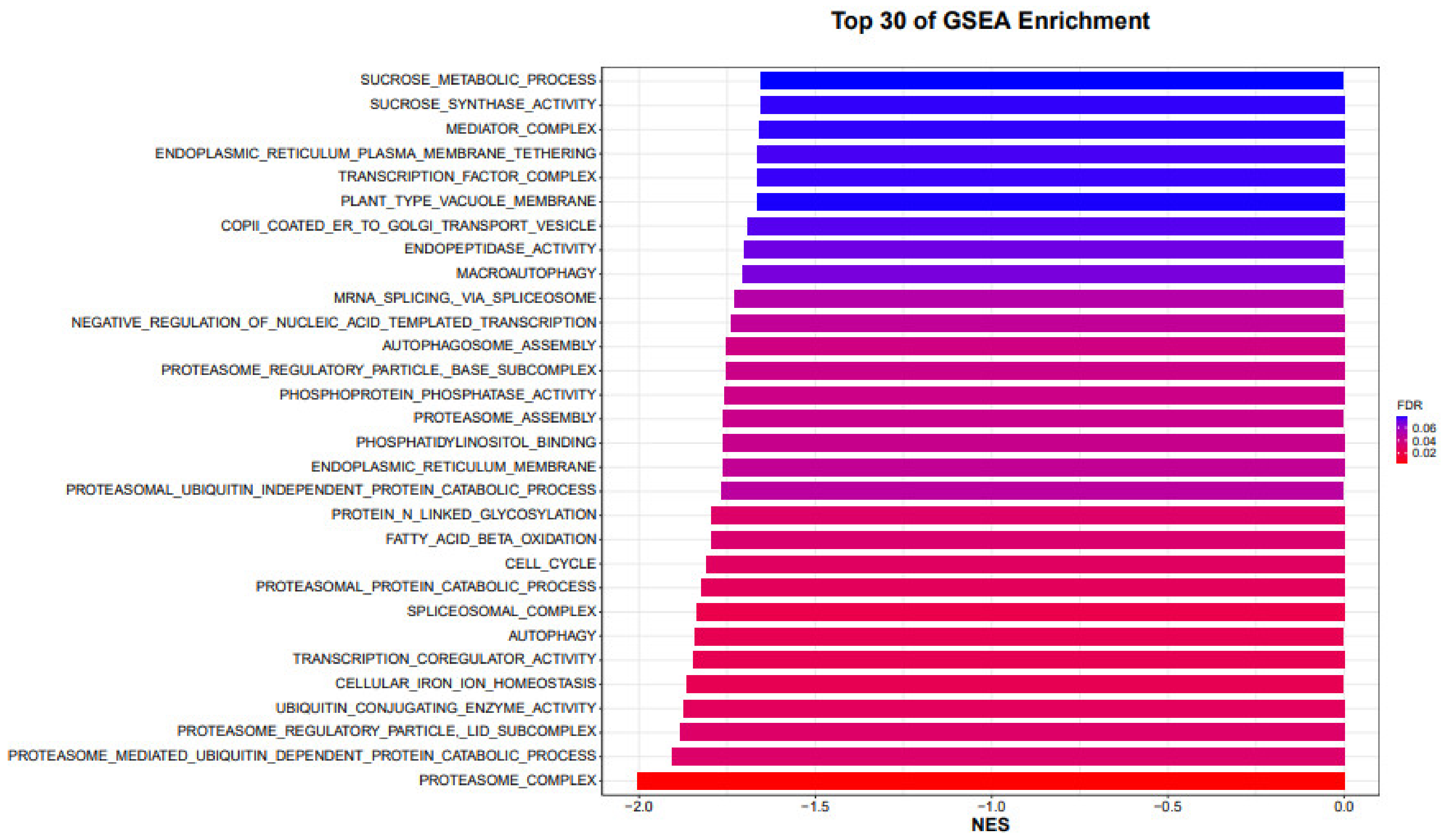
In summary, H21 was able to accumulate more solvent sugars in leaves and keep up with the consistence of photosynthesis during dry conditions, which resulted in it being more drought resistant than 478. The contribution of characteristics associated to the biosynthesis of cell walls to the preservation of water in H21. The incorporation of a synchronized multienzyme complex in the process of polysaccharide production provided regions of stability for the combination of a cell wall (
Figure 8). The increased supply of glycosyltransferase contributed to the drought-induced cytokinin deficiency in
Arabidopsis thaliana freaks. The investigation of GO development revealed that in H21 a number of DEGs were associated with cell wall connection. This was notably the case with the development family, which was induced by a variety of abiotic stressors and ABA. It was observed that increasing stomatal expression levels decreased their thickness (
Figure 9).
Moreover, expansions could decrease water misfortune by ceasing cell wall movement and hardening cell structures [
15]. There was a strong correlation between drought resistance in maize and both the lignin concentration and the middle outcome of the phenylpropanoid mix route (
Figure 11). The accumulation of caffeic abrasive and p-coumaric abrasive in the xylem sap influenced the growth of maize leaves during periods of drought by having an effect on lignin production. The abrasive effects of p-coumaric acid might rapidly lignify tissues when placed under strain and further enhance adaptability when exposed to drought. In the most recent study, KEGG and GO enhancement studies revealed that lignin-related metabolic reactions, such as the phenylpropanoid blend route, were improved in H21 under drought conditions. This was shown to be the case in the plant. The most likely scenario is that the phenylpropanoid production pathway and its components, including caffeic corrosive and p-coumaric corrosive, were accountable for reaction of H21 to drought (
Figure 12).
Plants’ ability to withstand drought is a complicated process with varied responses. At the molecular scale, as well as the cellular scale, a wide array of proteins are engaged in the process of adapting to and responding to drought. The genes that code for functional proteins and the genes that regulate those proteins may be used to classify these genetic traits [
9]. There are many different types of structural proteins, some of which include transporters, detoxifying enzymes, osmolyte biosynthesis enzymes, late embryogenesis abundant (LEA) proteins, and others [
15]. In order to regulate transcription factors, a regulatory network configuration that incorporates stress detection, cell signaling, and functional gene regulation has been designed [
16].
A major objective of the study was to demonstrate that classifying maize lines using high-density molecular marker data is a precise approach that does not require extensive field-combining ability studies [
8]. The current study’s findings are consistent with past genetic diversity studies that have found SNPs to be a valuable tool for inbred grouping lines into genetically related groups and for guiding hybrid crossings between members of various groups to provide superior yield performance. SNPs are also helpful for genotyping and integrating new inbred lines in a new cluster analysis, which can help to breed by swiftly detecting the genetic link of new inbred lines. Trace endogenous substances come in many forms as plant hormones. The loss in plant growth under drought-stress circumstances may exist due to an altered hormonal balance. Ultra-trace levels of plant hormones might play a crucial role in plant development, growth, and fast response to abiotic and biotic stressors. The Brassinosteroid C-6 Oxidase, which catalyzes the last steps of brassinosteroid production, is encoded by the Brassinosteroid-deficient dwarf1 gene. It is also interestingly linked to documented height loci in maize. Plants that overexpress ABA, which is necessary for drought resistance, show improved drought tolerance. However, ABA 8′-hydroxylase hydroxylates ABA to 8′-hydroxy- abscisate and NADP
+, depleting ABA to lower ABA levels and resulting in phenotypes ABA-deficient.
Comparing the categorization of maize lines conducted earlier using pedigree and breeding data to the classification achieved using SNPs, revealed good accuracy. Although the majority of the genotypes in this research fell into one of the four heterotic categories that the company’s breeding program had previously developed, there were be seen, demonstrating the large amount of genetic diversity present in maize germplasm [
9]. Furthermore, the genotyping information derived from this work may be used to further hybrid prediction models, allowing for more precise identification of hybrids, and increasing breeding effectiveness [
12].



 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}