

The Complete Chloroplast Genome of An Ophiorrhiza baviensis Drake Species Reveals Its Molecular Structure, Comparative, and Phylogenetic Relationships

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Chloroplast Genome Sequencing
2.2. DNA Extraction and Chloroplast Genome Sequencing
2.3. Genome Assembly and Annotation
2.4. Genome Comparison and Phylogenetic Identification
3. Results
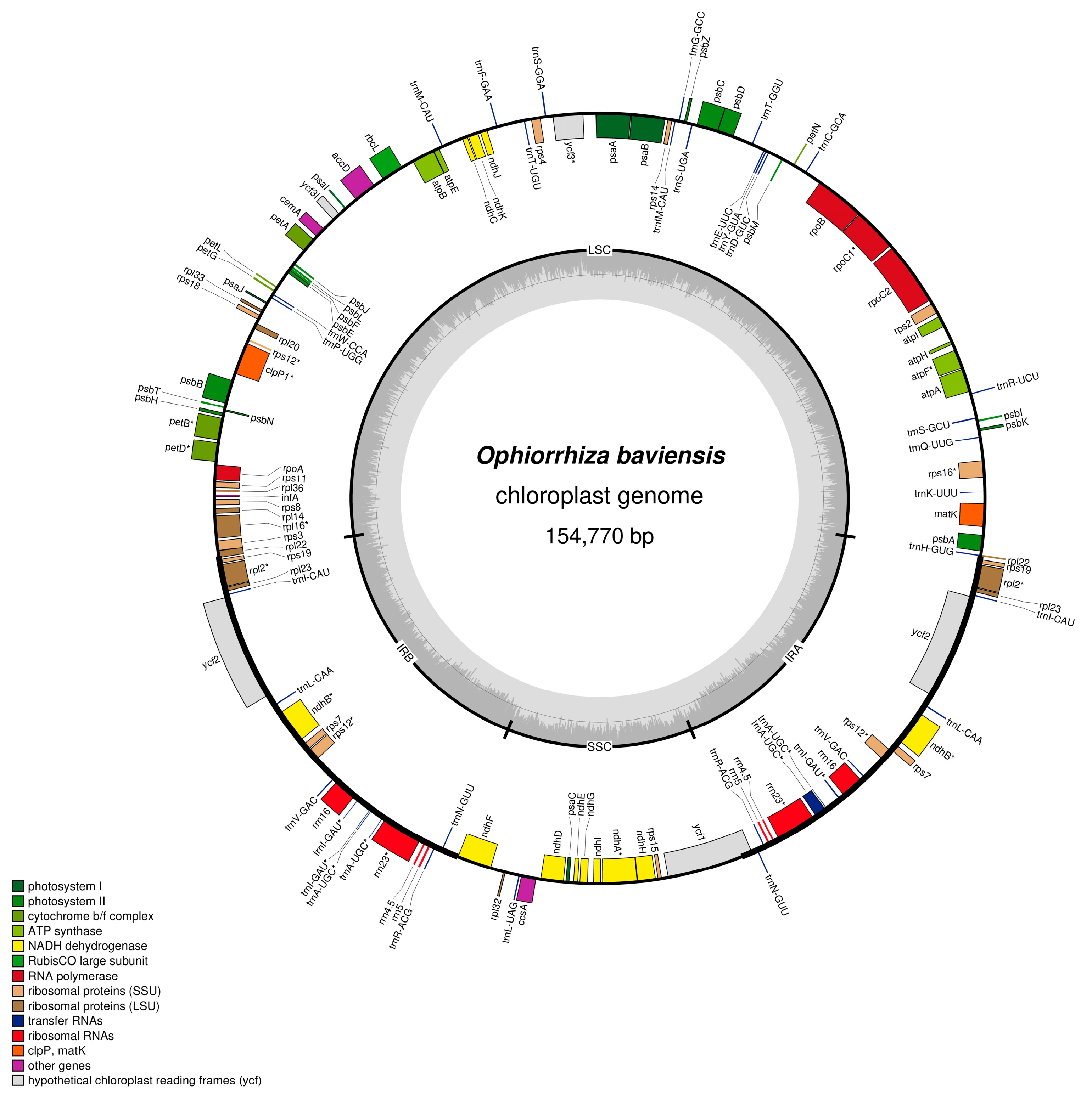
3.1. Chloroplast Genome Assembly and Annotation
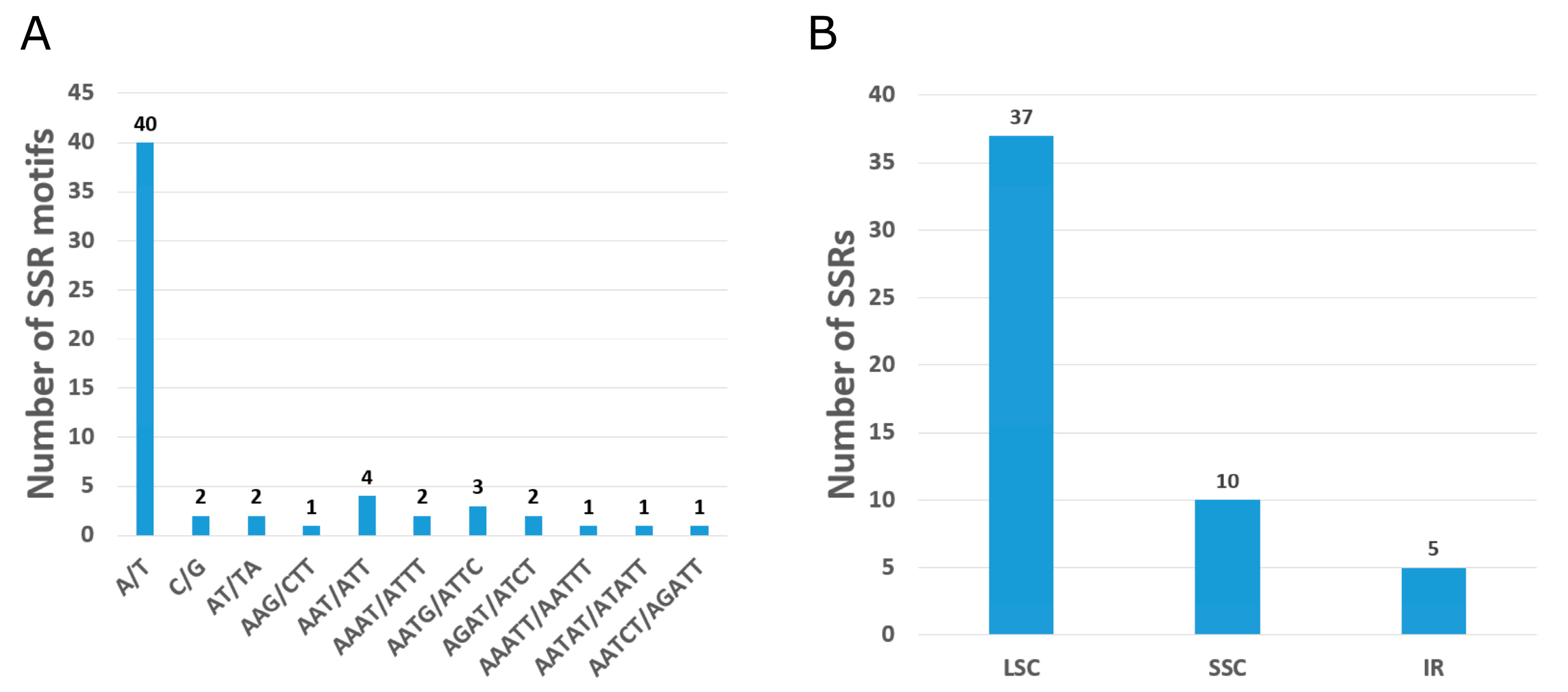
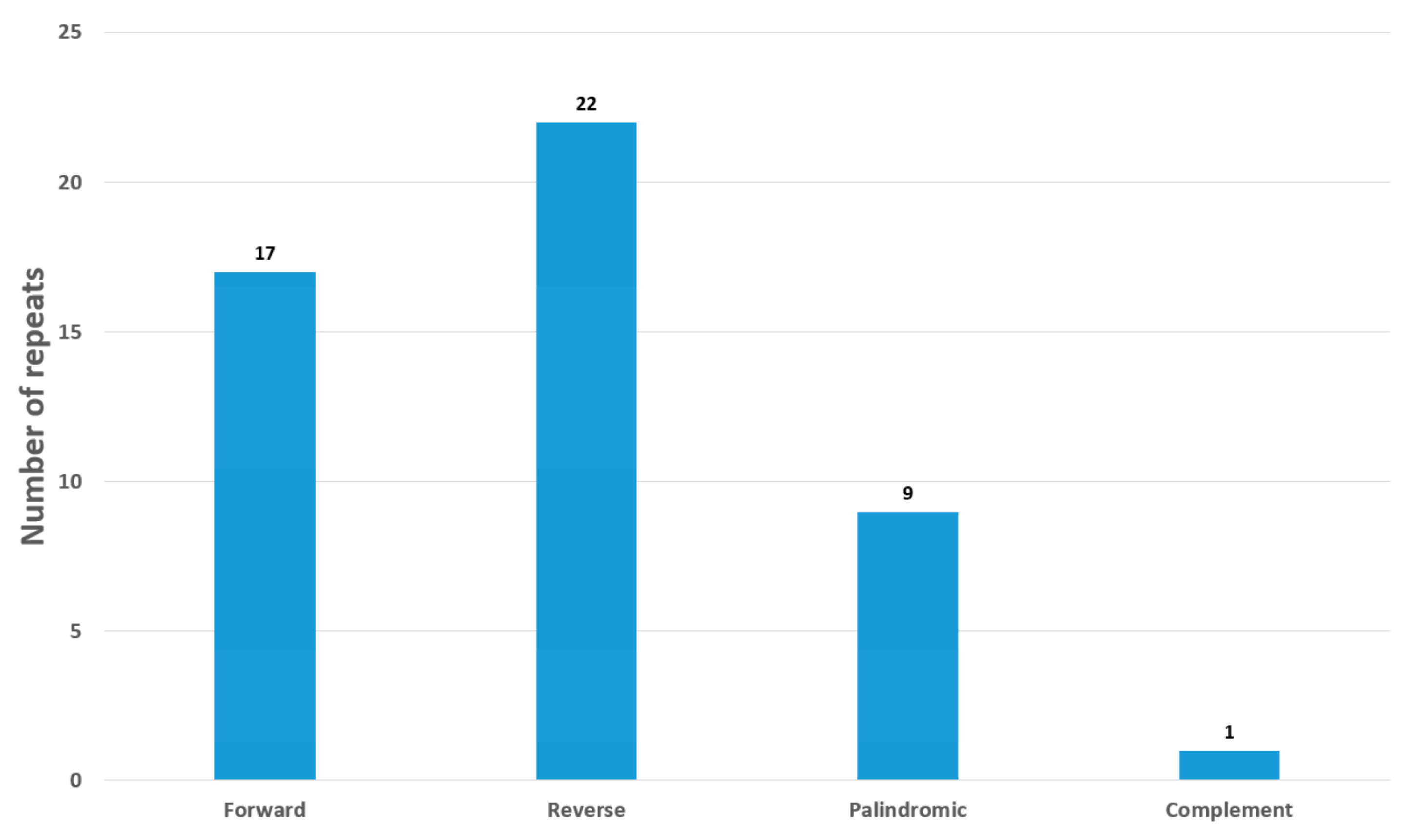
3.2. Repeat Sequences and Codon Analysis
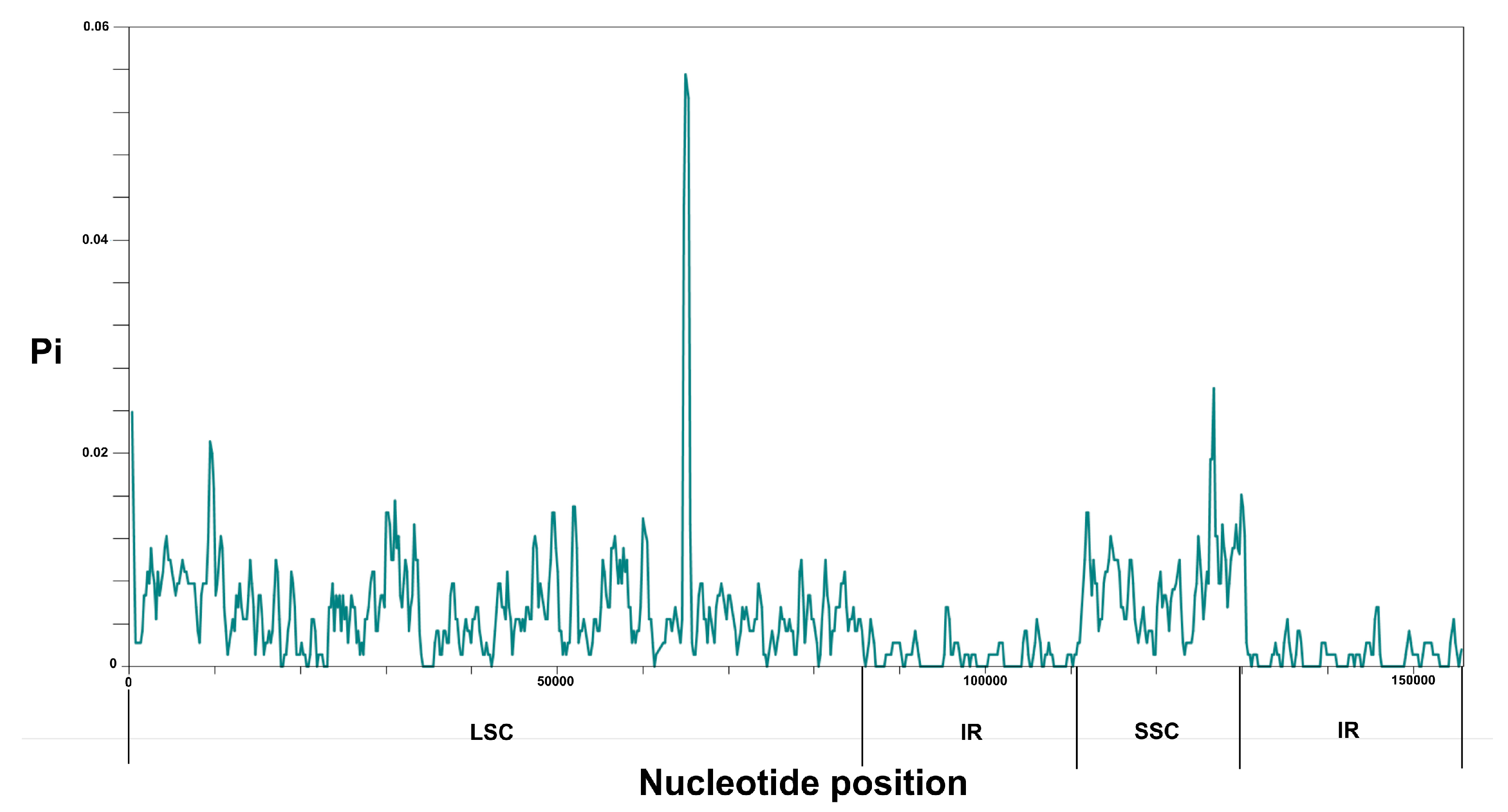
3.3. Chloroplast Genome Comparison
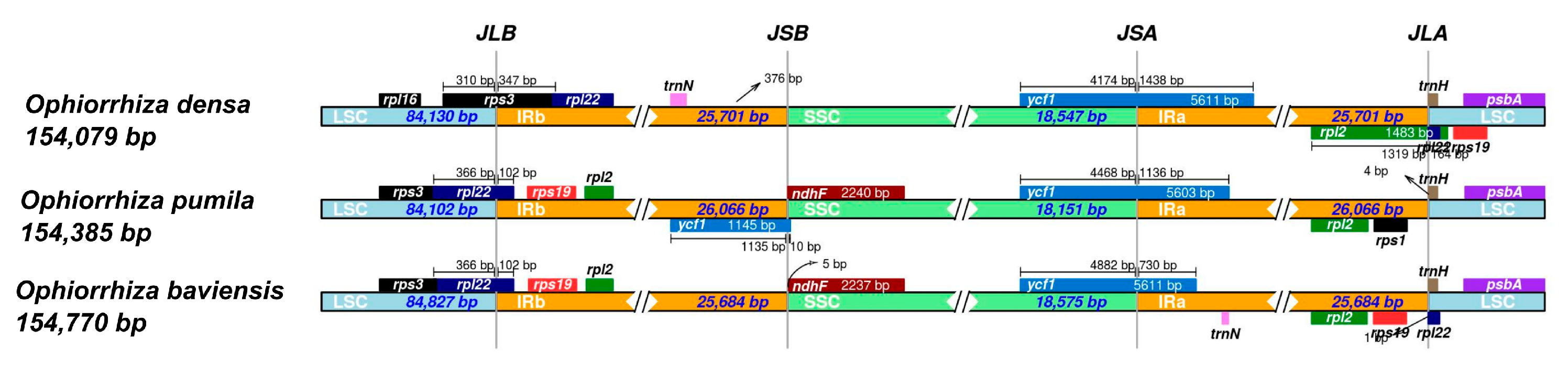
3.4. IR Contraction and Expansion in the Chloroplast Genome
3.5. Phylogenetic Inference
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic Metabolism in Plastids. Annu. Rev. Plant Biol. 2000, 51, 111. [Google Scholar] [CrossRef]
- Bendich, A.J. Circular Chloroplast Chromosomes: The Grand Illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef]
- Lei, W.; Liu, W.-J.; Nguyen, K.S. Revision of Three Taxa of Ophiorrhiza (Rubiaceae) from China. Phytotaxa 2019, 387, 129–139. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- PacificBiosciences. Pbmm2: A Minimap2 Frontend for PacBio Native Data Formats. Available online: https://github.com/PacificBiosciences/pbmm2 (accessed on 10 January 2021).
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, Finished Microbial Genome Assemblies from Long-Read SMRT Sequencing Data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. TRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A Tool for the Easy Generation of High-Quality Custom Graphical Maps of Plastid and Mitochondrial Genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast Computation of Maximal Repeats in Complete Genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Dehal, P.; Arkin, A. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Chen, T.; Taylor, C. Ophiorrhiza. In Flora of China; Press, S., Ed.; Beijing & Missouri Botanical Garden Press: St. Louis, MO, USA, 2011; Volume 19, pp. 258–282. [Google Scholar]
- Lei, W.; Tan, Y.; Hareesh, V.S.; Liu, Q. Ophiorrhiza Macrocarpa (Rubiaceae), a New Viviparous Species from Yunnan, Southwestern China. Nord. J. Bot. 2018, 36, njb-01637. [Google Scholar] [CrossRef]
- Hamzah, A.S. Isolation, Characterization and Biological Activities of Chemical Constituents of Ophiorrhiza and Hedyotis Species. Ph.D. Dissertation, Universiti Pertanian Malaysia, Serdang, Malaysia, 1994. [Google Scholar]
- GAO, L.; SU, Y.-J.; WANG, T. Plastid Genome Sequencing, Comparative Genomics, and Phylogenomics: Current Status and Prospects. J. Syst. Evol. 2010, 48, 77–93. [Google Scholar] [CrossRef]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Coatney, C.G.; Bennetzen, J.L. Gene Loss and Genome Rearrangement in the Plastids of Five Hemiparasites in the Family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef]
- Oyebanji, O.; Zhang, R.; Chen, S.-Y.; Yi, T.-S. New Insights Into the Plastome Evolution of the Millettioid/Phaseoloid Clade (Papilionoideae, Leguminosae). Front. Plant Sci. 2020, 11, 151. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Sylvester, S.P.; Zang, M.; El-Kassaby, Y.A.; Fang, Y. Evolutionary Patterns of Nucleotide Substitution Rates in Plastid Genomes of Quercus. Ecol. Evol. 2021, 11, 13401–13414. [Google Scholar] [CrossRef]
- Abdullah; Mehmood, F.; Shahzadi, I.; Ali, Z.; Islam, M.; Naeem, M.; Mirza, B.; Lockhart, P.J.; Ahmed, I.; Waheed, M.T. Correlations among Oligonucleotide Repeats, Nucleotide Substitutions, and Insertion–Deletion Mutations in Chloroplast Genomes of Plant Family Malvaceae. J. Syst. Evol. 2021, 59, 388–402. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Li, M.; Xu, W.; Schwarzacher, T.; Heslop-Harrison, J.S. Comparative Chloroplast Genome Analyses of Avena: Insights into Evolutionary Dynamics and Phylogeny. BMC Plant Biol. 2020, 20, 406. [Google Scholar] [CrossRef] [PubMed]
- Ly, S.N.; Garavito, A.; De Block, P.; Asselman, P.; Guyeux, C.; Charr, J.-C.; Janssens, S.; Mouly, A.; Hamon, P.; Guyot, R. Chloroplast Genomes of Rubiaceae: Comparative Genomics and Molecular Phylogeny in Subfamily Ixoroideae. PLoS ONE 2020, 15, e0232295. [Google Scholar] [CrossRef] [PubMed]
- Amenu, S.G.; Wei, N.; Wu, L.; Oyebanji, O.; Hu, G.; Zhou, Y.; Wang, Q. Phylogenomic and Comparative Analyses of Coffeeae Alliance (Rubiaceae): Deep Insights into Phylogenetic Relationships and Plastome Evolution. BMC Plant Biol. 2022, 22, 88. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Sigmund, R.; Börner, A.; Korzun, V.; Stein, N.; Sorrells, M.E.; Langridge, P.; Graner, A. Interspecific Transferability and Comparative Mapping of Barley EST-SSR Markers in Wheat, Rye and Rice. Plant Sci. 2005, 168, 195–202. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.; Chen, Z.; Zhou, S. A Chloroplast Genomic Strategy for Designing Taxon Specific DNA Mini-Barcodes: A Case Study on Ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast Microsatellites: New Tools for Studies in Plant Ecology and Evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-H.; Liu, X.; Cui, Y.-X.; Nie, L.-P.; Lin, Y.-L.; Wei, X.-P.; Wang, Y.; Yao, H. Molecular Structure and Phylogenetic Analyses of the Complete Chloroplast Genomes of Three Original Species of Pyrrosiae Folium. Chin. J. Nat. Med. 2020, 18, 573–581. [Google Scholar] [CrossRef]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; Chen, S.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-Optimal Codon Usage Affects Expression, Structure and Function of Clock Protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef]
- Somaratne, Y.; Guan, D.-L.; Wang, W.-Q.; Zhao, L.; Xu, S.-Q. The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2020, 9, 51. [Google Scholar] [CrossRef]
- Lyu, X.; Liu, Y. Nonoptimal Codon Usage Is Critical for Protein Structure and Function of the Master General Amino Acid Control Regulator CPC-1. Mbio 2020, 11, e02605-20. [Google Scholar] [CrossRef]
- Ding, S.; Dong, X.; Yang, J.; Guo, C.; Cao, B.; Guo, Y.; Hu, G. Complete Chloroplast Genome of Clethra Fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships. Forests 2021, 12, 441. [Google Scholar] [CrossRef]
- Kim, K.-J.; Lee, H.-L. Complete Chloroplast Genome Sequences from Korean Ginseng (Panax Schinseng Nees) and Comparative Analysis of Sequence Evolution among 17 Vascular Plants. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the Complete Chloroplast Genome into the Evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Lozano, J.J.; Ortiz, A.R. Comparative Analysis of Chloroplast Genomes: Functional Annotation, Genome-Based Phylogeny, and Deduced Evolutionary Patterns. Genome Res. 2002, 12, 567–583. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using Plastid Genome-Scale Data to Resolve Enigmatic Relationships among Basal Angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Razafimandimbison, S.G.; Rydin, C. Molecular-Based Assessments of Tribal and Generic Limits and Relationships in Rubiaceae (Gentianales): Polyphyly of Pomazoteae and Paraphyly of Ophiorrhizeae and Ophiorrhiza. Taxon 2019, 68, 72–91. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | O. baviensis Drake |
|---|---|
| Genome size (bp) | 154,770 bp |
| LSC size (bp) | 84,826 |
| SSC size (bp) | 18,574 |
| IR size (bp) | 25,685 |
| GC content (%) | 37.6 |
| No. of genes | 128 |
| No. of PCGs | 87 |
| No. of tRNA | 33 |
| No. of rRNA | 8 |
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| Photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpFa, atpH, atpI |
| Subunits of NADH-dehydrogenase | ndhAa, ndhB (×2)a, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petL, petB, petG, petA, petD, petN | |
| Subunits of photosystem I | psaJ, psaC, psaA, psaI, psaB | |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunit of rubisco | rbcL | |
| Transcription and translation | Large subunit of ribosome | rpl14, rpl16a, rpl2 (×2)a, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoB, rpoA, rpoC1a, rpoC2 | |
| Small subunit of ribosomal proteins | rps11, rps12(×2)a, rps14, rps15, rps16a, rps18, rps19 (×2), rps2, rps3, rps4, rps7 (×2), rps8 | |
| rRNA genes | rrn23S (×2), rrn16S (×2), rrn5S (×2), rrn4.5S (×2) | |
| tRNA genes | trnA-UGC (×2)a, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCCa, trnH-GUG, trnI-GAU (×2)a, trnL-CAA (×2), trnL-UAG, trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnW-CCA, trnY-GUA | |
| Translational initiation factor | infA | |
| Other genes | Subunit of acetyl-CoA-carboxylase (fatty acid synthesis) | accD |
| c-type cytochrome synthesis gene | ccsA | |
| Envelope membrane protein (carbon metabolism) | cemA | |
| Protease | clpPb | |
| Maturase (RNA processing) | matK | |
| Conserved open reading frames | ycf1, ycf2 (×2), ycf3b |
| Codon | AA | Frequency | RCSU | Codon | AA | Frequency | RCSU | Codon | AA | Frequency | RCSU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UAA | * | 1259 | 1.22 | AUC | I | 1205 | 0.80 | CGG | R | 420 | 0.75 |
| UAG | * | 825 | 0.80 | AUA | I | 1471 | 0.98 | AGA | R | 1093 | 1.94 |
| UGA | * | 1004 | 0.98 | AAA | K | 2050 | 1.35 | AGG | R | 627 | 1.12 |
| GCU | A | 446 | 1.23 | AAG | K | 982 | 0.65 | UCU | S | 1113 | 1.39 |
| GCC | A | 351 | 0.97 | UUA | L | 1040 | 1.23 | UCC | S | 982 | 1.22 |
| GCA | A | 401 | 1.11 | UUG | L | 1095 | 1.30 | UCA | S | 824 | 1.03 |
| GCG | A | 250 | 0.69 | CUU | L | 1063 | 1.26 | UCG | S | 622 | 0.77 |
| UGU | C | 679 | 1.20 | CUC | L | 653 | 0.77 | AGU | S | 747 | 0.93 |
| UGC | C | 449 | 0.80 | CUA | L | 737 | 0.87 | AGC | S | 529 | 0.66 |
| GAU | D | 1012 | 1.42 | CUG | L | 480 | 0.57 | ACU | T | 668 | 1.13 |
| GAC | D | 413 | 0.58 | AUG | M | 856 | 1.00 | ACC | T | 651 | 1.10 |
| GAA | E | 1337 | 1.43 | AAU | N | 1779 | 1.38 | ACA | T | 647 | 1.09 |
| GAG | E | 537 | 0.57 | AAC | N | 800 | 0.62 | ACG | T | 406 | 0.68 |
| UUU | F | 2212 | 1.20 | CCU | P | 611 | 1.03 | GUU | V | 784 | 1.38 |
| UUC | F | 1481 | 0.80 | CCC | P | 618 | 1.04 | GUC | V | 411 | 0.72 |
| GGU | G | 540 | 0.96 | CCA | P | 726 | 1.23 | GUA | V | 682 | 1.20 |
| GGC | G | 383 | 0.68 | CCG | P | 414 | 0.70 | GUG | V | 402 | 0.71 |
| GGA | G | 747 | 1.33 | CAA | Q | 987 | 1.40 | UGG | W | 681 | 1.00 |
| GGG | G | 577 | 1.03 | CAG | Q | 420 | 0.60 | UAU | Y | 1345 | 1.36 |
| CAU | H | 880 | 1.36 | CGU | R | 376 | 0.67 | UAC | Y | 637 | 0.64 |
| CAC | H | 414 | 0.64 | CGC | R | 280 | 0.50 | ||||
| AUU | I | 1830 | 1.22 | CGA | R | 576 | 1.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, M.H.; Tran, T.H.; Le, T.D.; Le, T.L.; Hoang, H.; Chu, H.H. The Complete Chloroplast Genome of An Ophiorrhiza baviensis Drake Species Reveals Its Molecular Structure, Comparative, and Phylogenetic Relationships. Genes 2023, 14, 227. https://doi.org/10.3390/genes14010227
Pham MH, Tran TH, Le TD, Le TL, Hoang H, Chu HH. The Complete Chloroplast Genome of An Ophiorrhiza baviensis Drake Species Reveals Its Molecular Structure, Comparative, and Phylogenetic Relationships. Genes. 2023; 14(1):227. https://doi.org/10.3390/genes14010227
Chicago/Turabian StylePham, Mai Huong, Thu Hoai Tran, Thi Dung Le, Tung Lam Le, Ha Hoang, and Hoang Ha Chu. 2023. "The Complete Chloroplast Genome of An Ophiorrhiza baviensis Drake Species Reveals Its Molecular Structure, Comparative, and Phylogenetic Relationships" Genes 14, no. 1: 227. https://doi.org/10.3390/genes14010227
APA StylePham, M. H., Tran, T. H., Le, T. D., Le, T. L., Hoang, H., & Chu, H. H. (2023). The Complete Chloroplast Genome of An Ophiorrhiza baviensis Drake Species Reveals Its Molecular Structure, Comparative, and Phylogenetic Relationships. Genes, 14(1), 227. https://doi.org/10.3390/genes14010227