
Evolutionary Landscape of SOX Genes to Inform Genotype-to-Phenotype Relationships
 ,
,  ,
,  , , , , ,
, , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence and Structure Analysis
2.2. Genomic Variant Analysis
2.3. SOX18 Culture Experiments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3. Results
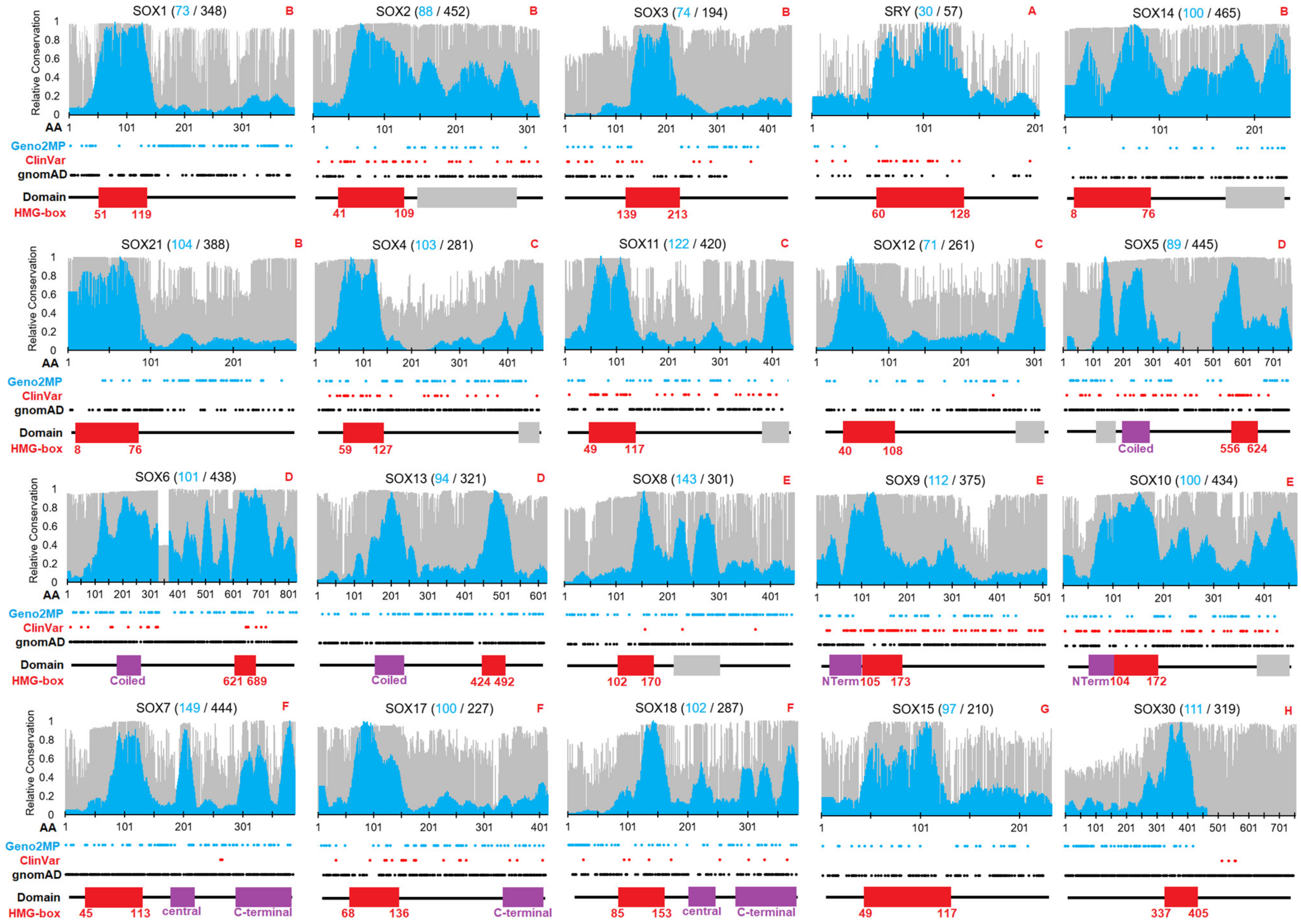
3.1. Sequence Evolution of SOX Genes/Proteins
| Gene | Group | Ensembl | NCBI | Missense Constraint | OMIM/ClinVar | Inheritance | AA | Species NA | Species AA | gnomAD | ClinVar | Geno2MP | COSMIC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SOX1 | B | ENST00000330949 | NP 005977 | 0.58 | - | - | 391 | 73 | 348 | 120 | 0 | 115 | 134 |
| SOX2 | B | ENST00000325404 | NP 003097 | 2.12 | Microphthalmia | AD | 317 | 88 | 452 | 119 | 70 | 66 | 203 |
| SOX3 | B | ENST00000370536 | NP 005625 | 2.21 | Panhypopituitarism | XL | 446 | 74 | 194 | 104 | 18 | 39 | 226 |
| SRY | A | ENST00000383070 | NP 003131 | −0.14 | sex reversal | XLD | 204 | 30 | 57 | 25 | 26 | 5 | 25 |
| SOX14 | B | ENST00000306087 | NP 004180 | 1.14 | - | - | 240 | 100 | 465 | 118 | 0 | 20 | 112 |
| SOX21 | B | ENST00000376945 | NP 009015 | 1.74 | - | - | 276 | 104 | 388 | 81 | 0 | 60 | 79 |
| SOX4 | C | ENST00000244745 | NP 003098 | 1.17 | Coffin-Siris syndrome | AD | 474 | 103 | 281 | 210 | 27 | 64 | 125 |
| SOX11 | C | ENST00000322002 | NP 003099 | 2.26 | Coffin-Siris syndrome | AD | 441 | 122 | 420 | 166 | 47 | 38 | 282 |
| SOX12 | C | ENST00000342665 | NP 008874 | 1.79 | - | - | 315 | 71 | 261 | 106 | 1 | 27 | 63 |
| SOX5 | D | ENST00000451604 | NP 008871 | 3.15 | Lamb-Shaffer syndrome | AD | 763 | 89 | 445 | 264 | 46 | 51 | 480 |
| SOX6 | D | ENST00000528429 | NP 001354802 | 1.94 | Tolchin-Le Caignec syndrome | AD | 828 | 101 | 438 | 365 | 17 | 86 | 380 |
| SOX13 | D | ENST00000367204 | NP 005677 | 1.74 | - | - | 622 | 94 | 321 | 296 | 0 | 91 | 199 |
| SOX8 | E | ENST00000293894 | NP 055402 | 0.89 | - | - | 446 | 143 | 301 | 254 | 3 | 98 | 183 |
| SOX9 | E | ENST00000245479 | NP 000337 | 1.63 | Campomelic dysplasia | AD | 509 | 112 | 375 | 259 | 97 | 55 | 524 |
| SOX10 | E | ENST00000396884 | NP 008872 | 2.77 | PCWH syndrome | AD | 466 | 100 | 434 | 176 | 100 | 44 | 198 |
| SOX7 | F | ENST0000030450 | NP 113627 | −0.74 | - | - | 388 | 149 | 444 | 420 | 2 | 81 | 204 |
| SOX17 | F | ENST00000297316 | NP 071899 | 0.77 | Vesicoureteral reflux | AD | 414 | 100 | 227 | 225 | 18 | 63 | 424 |
| SOX18 | F | ENST00000340356 | NP 060889 | 0.86 | HLTS | AD/AR | 384 | 102 | 287 | 151 | 9 | 75 | 93 |
| SOX15 | G | ENST00000250055 | NP 008873 | 0.62 | - | - | 233 | 97 | 210 | 118 | 0 | 29 | 83 |
| SOX30 | H | ENST00000265007 | NP 848511 | 0.78 | Male infertility | AD | 753 | 111 | 319 | 422 | 4 | 67 | 296 |
| Average | 1 | Total | 8910 | 1963 | 6667 | 3999 | 485 | 1174 | 4313 |
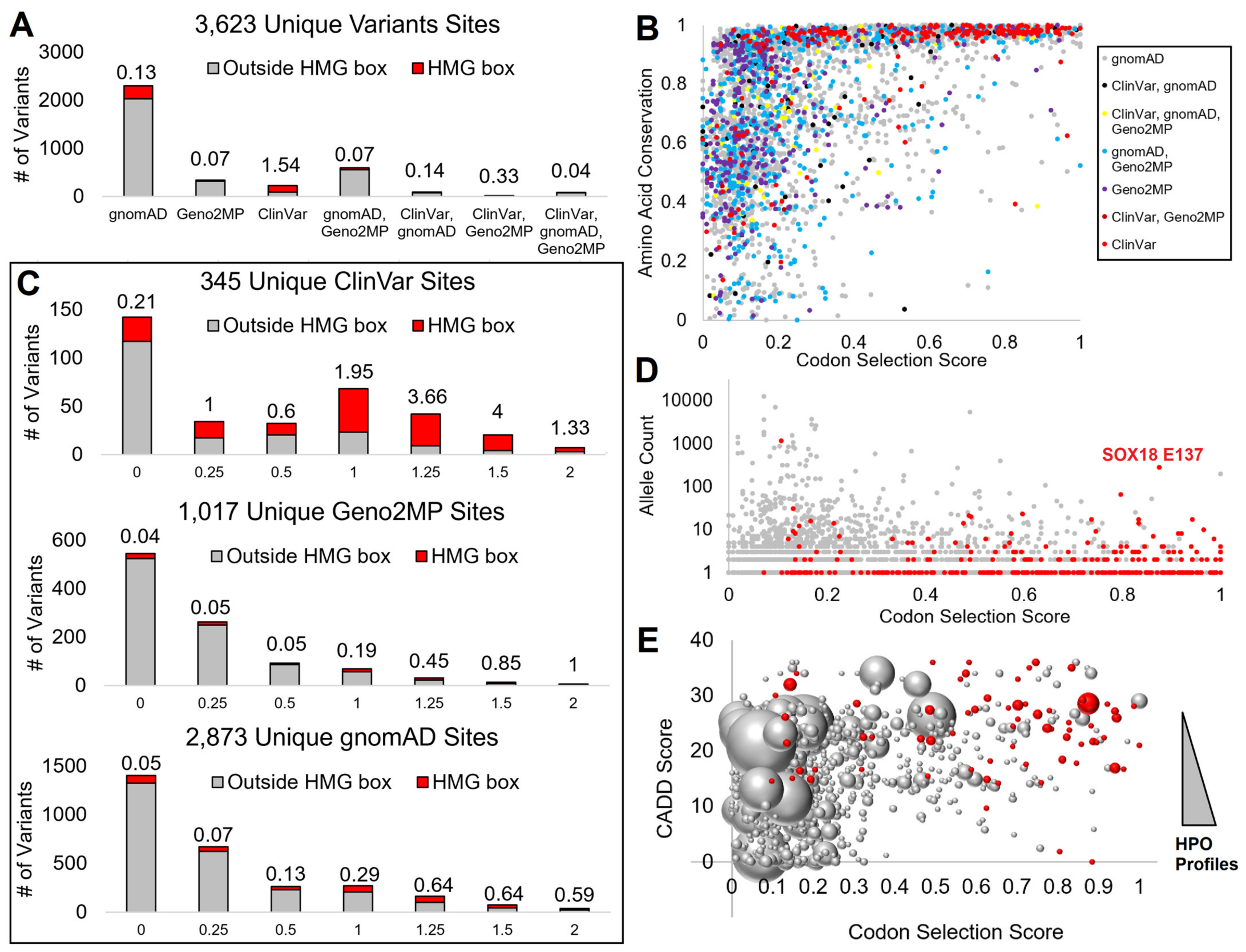
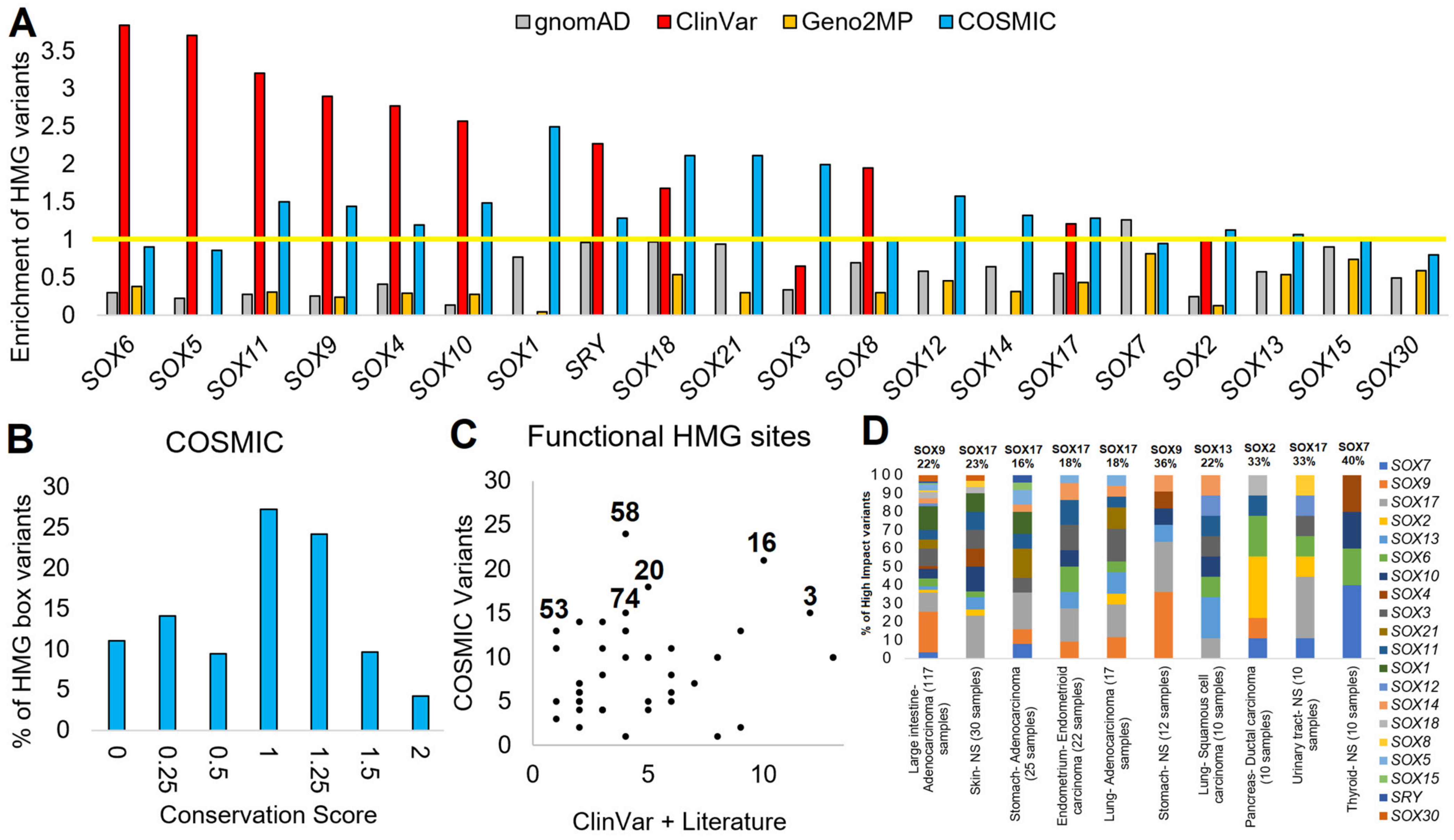
3.2. Mapping SOX Variants
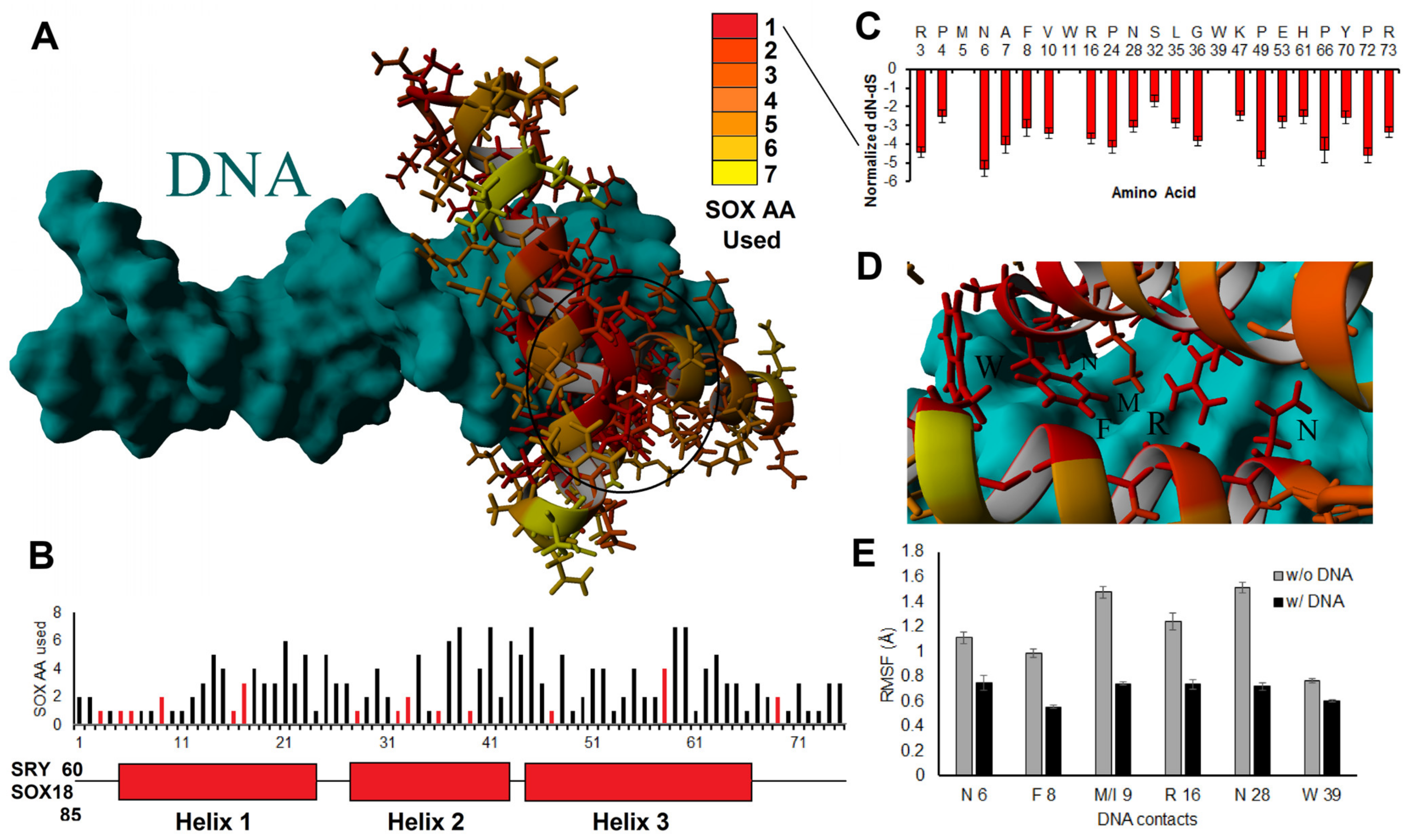
3.3. Interpreting HMG Box Variants
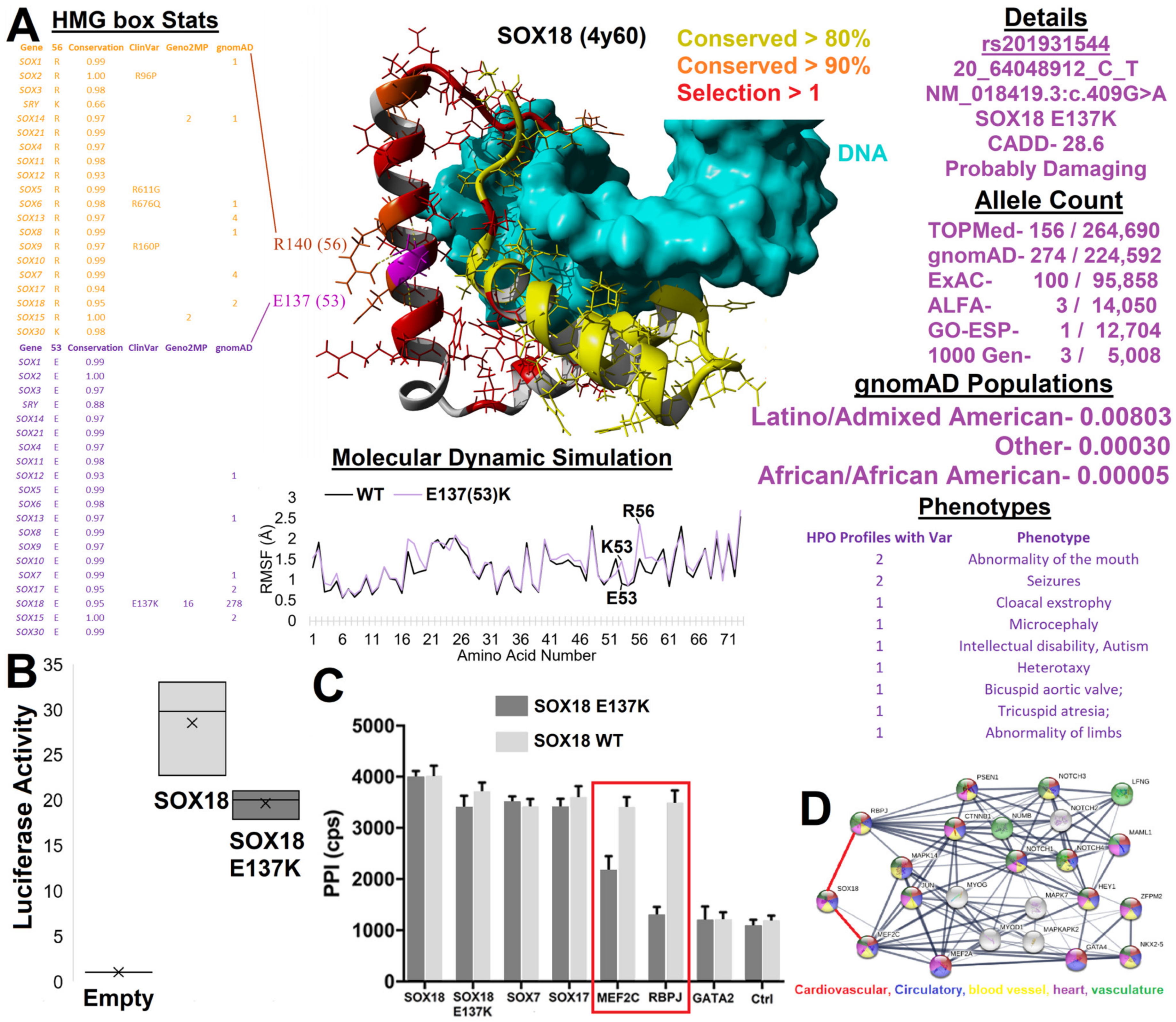
3.4. HMG Box Variant SOX18 E137K
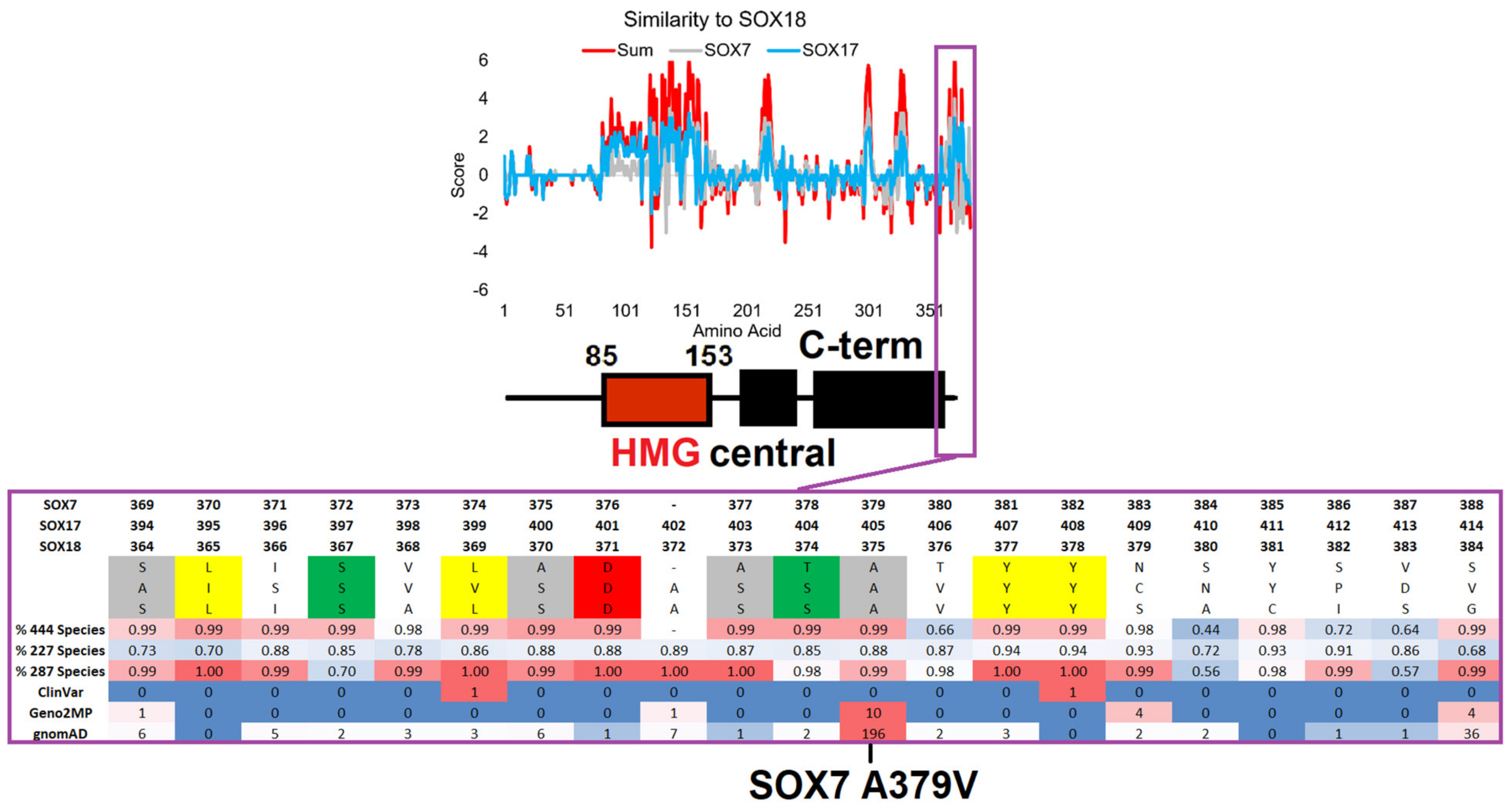
3.5. Functional SOX Variants Outside the HMG Box
3.6. Somatic SOX Variants in Cancer
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pevny, L.H.; Lovell-Badge, R. Sox genes find their feet. Curr. Opin. Genet. Dev. 1997, 7, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef]
- Goodfellow, P.N.; Lovell-Badge, R. SRY and sex determination in mammals. Annu. Rev. Genet. 1993, 27, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.C. Back to basics: Sox genes. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 2356–2366. [Google Scholar] [CrossRef]
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX Family of Developmental Transcription Factors Based on Sequence and Structural Indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Pontiggia, A.; Whitfield, S.; Goodfellow, P.N.; Lovell-Badge, R.; Bianchi, M.E. Evolutionary conservation in the DNA-binding and -bending properties of HMG-boxes from SRY proteins of primates. Gene 1995, 154, 277–280. [Google Scholar] [CrossRef]
- Read, C.M.; Cary, P.D.; Preston, N.S.; Lnenicek-Allen, M.; Crane-Robinson, C. The DNA sequence specificity of HMG boxes lies in the minor wing of the structure. EMBO J. 1994, 13, 5639–5646. [Google Scholar] [CrossRef]
- Murphy, E.C.; Zhurkin, V.B.; Louis, J.M.; Cornilescu, G.; Clore, G.M. Structural basis for SRY-dependent 46-X,Y sex reversal: Modulation of DNA bending by a naturally occurring point mutation. J. Mol. Biol. 2001, 312, 481–499. [Google Scholar] [CrossRef]
- Williams, D.C., Jr.; Cai, M.; Clore, G.M. Molecular basis for synergistic transcriptional activation by Oct1 and Sox2 revealed from the solution structure of the 42-kDa Oct1.Sox2.Hoxb1-DNA ternary transcription factor complex. J. Biol. Chem. 2004, 279, 1449–1457. [Google Scholar] [CrossRef]
- Hawkins, J.R.; Taylor, A.; Berta, P.; Levilliers, J.; Van der Auwera, B.; Goodfellow, P.N. Mutational analysis of SRY: Nonsense and missense mutations in XY sex reversal. Hum. Genet. 1992, 88, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Fantes, J.; Ragge, N.K.; Lynch, S.-A.; McGill, N.I.; Collin, J.R.O.; Howard-Peebles, P.N.; Hayward, C.; Vivian, A.J.; Williamson, K.; van Heyningen, V.; et al. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003, 33, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, S.A.; Pauer, G.J.T.; Reid, J.; Simpson, E.; Crowe, S.; Maumenee, I.H.; Traboulsi, E.I. SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. Am. J. Med. Genet. A 2005, 138A, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.S.; Cundall, M.; Turton, J.; Rizotti, K.; Mehta, A.; Palmer, R.; Wong, J.; Chong, W.K.; Al-Zyoud, M.; El-Ali, M.; et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am. J. Hum. Genet. 2005, 76, 833–849. [Google Scholar] [CrossRef]
- Pingault, V.; Bondurand, N.; Kuhlbrodt, K.; Goerich, D.E.; Préhu, M.O.; Puliti, A.; Herbarth, B.; Hermans-Borgmeyer, I.; Legius, E.; Matthijs, G.; et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 1998, 18, 171–173. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Koshimizu, E.; Ohashi, H.; Phadke, S.; Kou, I.; Shiina, M.; Suzuki, T.; Okamoto, N.; Imamura, S.; Yamashita, M.; et al. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat. Commun. 2014, 5, 4011. [Google Scholar] [CrossRef]
- Irrthum, A.; Devriendt, K.; Chitayat, D.; Matthijs, G.; Glade, C.; Steijlen, P.M.; Fryns, J.-P.; Van Steensel, M.A.M.; Vikkula, M. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am. J. Hum. Genet. 2003, 72, 1470–1478. [Google Scholar] [CrossRef]
- Angelozzi, M.; Lefebvre, V. SOXopathies: Growing Family of Developmental Disorders Due to SOX Mutations. Trends Genet. TIG 2019, 35, 658–671. [Google Scholar] [CrossRef]
- Srivastava, Y.; Tan, D.S.; Malik, V.; Weng, M.; Javed, A.; Cojocaru, V.; Wu, G.; Veerapandian, V.; Cheung, L.W.T.; Jauch, R. Cancer-associated missense mutations enhance the pluripotency reprogramming activity of OCT4 and SOX17. FEBS J. 2020, 287, 122–144. [Google Scholar] [CrossRef]
- Prokop, J.W.; Leeper, T.C.; Duan, Z.-H.; Milsted, A. Amino acid function and docking site prediction through combining disease variants, structure alignments, sequence alignments, and molecular dynamics: A study of the HMG domain. BMC Bioinform. 2012, 13 (Suppl. S2), S3. [Google Scholar] [CrossRef]
- Dong, C.; Wilhelm, D.; Koopman, P. Sox genes and cancer. Cytogenet. Genome Res. 2004, 105, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Lazar, J.; Crapitto, G.; Smith, D.C.; Worthey, E.A.; Jacob, H.J. Molecular modeling in the age of clinical genomics, the enterprise of the next generation. J. Mol. Model. 2017, 23, 75. [Google Scholar] [CrossRef]
- Prokop, J.W.; Jdanov, V.; Savage, L.; Morris, M.; Lamb, N.; VanSickle, E.; Stenger, C.L.; Rajasekaran, S.; Bupp, C.P. Computational and Experimental Analysis of Genetic Variants. Compr. Physiol. 2022, 12, 3303–3336. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [PubMed]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinform. Oxf. Engl. 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Dinkel, H.; Michael, S.; Weatheritt, R.J.; Davey, N.E.; Van Roey, K.; Altenberg, B.; Toedt, G.; Uyar, B.; Seiler, M.; Budd, A.; et al. ELM—The database of eukaryotic linear motifs. Nucleic Acids Res. 2012, 40, D242–D251. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. S9), 114–122. [Google Scholar] [CrossRef]
- Fontaine, F.R.; Goodall, S.; Prokop, J.W.; Howard, C.B.; Moustaqil, M.; Kumble, S.; Rasicci, D.T.; Osborne, G.W.; Gambin, Y.; Sierecki, E.; et al. Functional domain analysis of SOX18 transcription factor using a single-chain variable fragment-based approach. mAbs 2018, 10, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Klaus, M.; Prokoph, N.; Girbig, M.; Wang, X.; Huang, Y.-H.; Srivastava, Y.; Hou, L.; Narasimhan, K.; Kolatkar, P.R.; Francois, M.; et al. Structure and decoy-mediated inhibition of the SOX18/Prox1-DNA interaction. Nucleic Acids Res. 2016, 44, 3922–3935. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Wang, J.; Al-Ouran, R.; Hu, Y.; Kim, S.-Y.; Wan, Y.-W.; Wangler, M.F.; Yamamoto, S.; Chao, H.-T.; Comjean, A.; Mohr, S.E.; et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet. 2017, 100, 843–853. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Prokop, J.W.; Tsaih, S.-W.; Faber, A.B.; Boehme, S.; Underwood, A.C.; Troyer, S.; Playl, L.; Milsted, A.; Turner, M.E.; Ely, D.; et al. The phenotypic impact of the male-specific region of chromosome-Y in inbred mating: The role of genetic variants and gene duplications in multiple inbred rat strains. Biol. Sex Differ. 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Stevers, L.M.; Giles, N.; Polinkovsky, M.E.; Moustaqil, M.; Mureev, S.; Johnston, W.A.; Dahmer-Heath, M.; Skalamera, D.; Gonda, T.J.; et al. Rapid mapping of interactions between Human SNX-BAR proteins measured in vitro by AlphaScreen and single-molecule spectroscopy. Mol. Cell. Proteom. 2014, 13, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Giles, N.; Polinkovsky, M.; Moustaqil, M.; Alexandrov, K.; Gambin, Y. A cell-free approach to accelerate the study of protein-protein interactions in vitro. Interface Focus 2013, 3, 20130018. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Gimelli, G.; Gimelli, S.; Dimasi, N.; Bocciardi, R.; Di Battista, E.; Pramparo, T.; Zuffardi, O. Identification and molecular modelling of a novel familial mutation in the SRY gene implicated in the pure gonadal dysgenesis. Eur. J. Hum. Genet. 2007, 15, 76–80. [Google Scholar] [CrossRef]
- Domenice, S.; Yumie Nishi, M.; Correia Billerbeck, A.E.; Latronico, A.C.; Aparecida Medeiros, M.; Russell, A.J.; Vass, K.; Marino Carvalho, F.; Costa Frade, E.M.; Prado Arnhold, I.J.; et al. A novel missense mutation (S18N) in the 5’ non-HMG box region of the SRY gene in a patient with partial gonadal dysgenesis and his normal male relatives. Hum. Genet. 1998, 102, 213–215. [Google Scholar] [CrossRef]
- Canto, P.; de la Chesnaye, E.; López, M.; Cervantes, A.; Chávez, B.; Vilchis, F.; Reyes, E.; Ulloa-Aguirre, A.; Kofman-Alfaro, S.; Méndez, J.P. A mutation in the 5’ non-high mobility group box region of the SRY gene in patients with Turner syndrome and Y mosaicism. J. Clin. Endocrinol. Metab. 2000, 85, 1908–1911. [Google Scholar] [CrossRef][Green Version]
- Goji, K.; Nishijima, E.; Tsugawa, C.; Nishio, H.; Pokharel, R.K.; Matsuo, M. Novel missense mutation in the HMG box of SOX9 gene in a Japanese XY male resulted in campomelic dysplasia and severe defect in masculinization. Hum. Mutat. 1998, 11 (Suppl. S1), S114–S116. [Google Scholar] [CrossRef]
- Kwok, C.; Weller, P.A.; Guioli, S.; Foster, J.W.; Mansour, S.; Zuffardi, O.; Punnett, H.H.; Dominguez-Steglich, M.A.; Brook, J.D.; Young, I.D. Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am. J. Hum. Genet. 1995, 57, 1028–1036. [Google Scholar]
- Scherer, G.; Held, M.; Erdel, M.; Meschede, D.; Horst, J.; Lesniewicz, R.; Midro, A.T. Three novel SRY mutations in XY gonadal dysgenesis and the enigma of XY gonadal dysgenesis cases without SRY mutations. Cytogenet. Cell Genet. 1998, 80, 188–192. [Google Scholar] [CrossRef]
- Meyer, J.; Südbeck, P.; Held, M.; Wagner, T.; Schmitz, M.L.; Bricarelli, F.D.; Eggermont, E.; Friedrich, U.; Haas, O.A.; Kobelt, A.; et al. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: Lack of genotype/phenotype correlations. Hum. Mol. Genet. 1997, 6, 91–98. [Google Scholar] [CrossRef]
- Braun, A.; Kammerer, S.; Cleve, H.; Löhrs, U.; Schwarz, H.P.; Kuhnle, U. True hermaphroditism in a 46,XY individual, caused by a postzygotic somatic point mutation in the male gonadal sex-determining locus (SRY): Molecular genetics and histological findings in a sporadic case. Am. J. Hum. Genet. 1993, 52, 578–585. [Google Scholar] [PubMed]
- Staffler, A.; Hammel, M.; Wahlbuhl, M.; Bidlingmaier, C.; Flemmer, A.W.; Pagel, P.; Nicolai, T.; Wegner, M.; Holzinger, A. Heterozygous SOX9 mutations allowing for residual DNA-binding and transcriptional activation lead to the acampomelic variant of campomelic dysplasia. Hum. Mutat. 2010, 31, E1436–E1444. [Google Scholar] [CrossRef] [PubMed]
- Affara, N.A.; Chalmers, I.J.; Ferguson-Smith, M.A. Analysis of the SRY gene in 22 sex-reversed XY females identifies four new point mutations in the conserved DNA binding domain. Hum. Mol. Genet. 1993, 2, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Chaoui, A.; Watanabe, Y.; Touraine, R.; Baral, V.; Goossens, M.; Pingault, V.; Bondurand, N. Identification and functional analysis of SOX10 missense mutations in different subtypes of Waardenburg syndrome. Hum. Mutat. 2011, 32, 1436–1449. [Google Scholar] [CrossRef]
- Haqq, C.M.; King, C.Y.; Ukiyama, E.; Falsafi, S.; Haqq, T.N.; Donahoe, P.K.; Weiss, M.A. Molecular basis of mammalian sexual determination: Activation of Müllerian inhibiting substance gene expression by SRY. Science 1994, 266, 1494–1500. [Google Scholar] [CrossRef]
- Berta, P.; Hawkins, J.R.; Sinclair, A.H.; Taylor, A.; Griffiths, B.L.; Goodfellow, P.N.; Fellous, M. Genetic evidence equating SRY and the testis-determining factor. Nature 1990, 348, 448–450. [Google Scholar] [CrossRef]
- Zeng, Y.T.; Ren, Z.R.; Zhang, M.L.; Huang, Y.; Zeng, F.Y.; Huang, S.Z. A new de novo mutation (A113T) in HMG box of the SRY gene leads to XY gonadal dysgenesis. J. Med. Genet. 1993, 30, 655–657. [Google Scholar] [CrossRef][Green Version]
- Schmitt-Ney, M.; Thiele, H.; Kaltwasser, P.; Bardoni, B.; Cisternino, M.; Scherer, G. Two novel SRY missense mutations reducing DNA binding identified in XY females and their mosaic fathers. Am. J. Hum. Genet. 1995, 56, 862–869. [Google Scholar]
- Lundberg, Y.; Ritzén, M.; Harlin, J.; Wedell, A. Novel missense mutation (P131R) in the HMG box of SRY in XY sex reversal. Hum. Mutat. 1998, 11, S328–S329. [Google Scholar] [CrossRef]
- Fernandez, R.; Marchal, J.A.; Sanchez, A.; Pasaro, E. A point mutation, R59G, within the HMG-SRY box in a female 45,X/46,X, psu dic(Y)(pter-->q11::q11-->pter). Hum. Genet. 2002, 111, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Vilain, E.; McElreavey, K.; Jaubert, F.; Raymond, J.P.; Richaud, F.; Fellous, M. Familial case with sequence variant in the testis-determining region associated with two sex phenotypes. Am. J. Hum. Genet. 1992, 50, 1008–1011. [Google Scholar] [PubMed]
- Hiort, O.; Gramss, B.; Klauber, G.T. True hermaphroditism with 46,XY karyotype and a point mutation in the SRY gene. J. Pediatr. 1995, 126, 1022. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Nishimura, G.; Nagai, T.; Sawai, H.; Yoshikata, M.; Miyagawa, S.; Hanita, T.; Sato, S.; Hasegawa, T.; Ishikawa, S.; et al. Mutation analysis of SOX9 and single copy number variant analysis of the upstream region in eight patients with campomelic dysplasia and acampomelic campomelic dysplasia. Am. J. Med. Genet. A 2009, 149A, 2882–2885. [Google Scholar] [CrossRef]
- Imai, A.; Takagi, A.; Tamaya, T. A novel sex-determining region on Y (SRY) missense mutation identified in a 46,XY female and also in the father. Endocr. J. 1999, 46, 735–739. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okuhara, K.; Tajima, T.; Nakae, J.; Fujieda, K. A novel missense mutation in the HMG box region of the SRY gene in a Japanese patient with an XY sex reversal. J. Hum. Genet. 2000, 45, 112–114. [Google Scholar] [CrossRef]
- Cunha, J.L.; Soardi, F.C.; Bernardi, R.D.; Oliveira, L.E.C.; Benedetti, C.E.; Guerra-Junior, G.; Maciel-Guerra, A.T.; de Mello, M.P. The novel p.E89K mutation in the SRY gene inhibits DNA binding and causes the 46,XY disorder of sex development. Braz. J. Med. Biol. Res. 2011, 44, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.R.; Taylor, A.; Goodfellow, P.N.; Migeon, C.J.; Smith, K.D.; Berkovitz, G.D. Evidence for increased prevalence of SRY mutations in XY females with complete rather than partial gonadal dysgenesis. Am. J. Hum. Genet. 1992, 51, 979–984. [Google Scholar]
- Dörk, T.; Stuhrmann, M.; Miller, K.; Schmidtke, J. Independent observation of SRY mutation I90M in a patient with complete gonadal dysgenesis. Hum. Mutat. 1998, 11, 90–91. [Google Scholar] [CrossRef]
- Maier, E.M.; Leitner, C.; Löhrs, U.; Kuhnle, U. True hermaphroditism in an XY individual due to a familial point mutation of the SRY gene. J. Pediatr. Endocrinol. Metab. 2003, 16, 575–580. [Google Scholar] [CrossRef]
- Schäffler, A.; Barth, N.; Winkler, K.; Zietz, B.; Rümmele, P.; Knüchel, R.; Schölmerich, J.; Palitzsch, K.D. Identification of a new missense mutation (Gly95Glu) in a highly conserved codon within the high-mobility group box of the sex-determining region Y gene: Report on a 46,XY female with gonadal dysgenesis and yolk-sac tumor. J. Clin. Endocrinol. Metab. 2000, 85, 2287–2292. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jakubiczka, S.; Bettecken, T.; Stumm, M.; Neulen, J.; Wieacker, P. Another mutation within the HMG-box of the SRY gene associated with Swyer syndrome. Hum. Mutat. 1999, 13, 85. [Google Scholar] [CrossRef]
- Jäger, R.J.; Harley, V.R.; Pfeiffer, R.A.; Goodfellow, P.N.; Scherer, G. A familial mutation in the testis-determining gene SRY shared by both sexes. Hum. Genet. 1992, 90, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Preiss, S.; Argentaro, A.; Clayton, A.; John, A.; Jans, D.A.; Ogata, T.; Nagai, T.; Barroso, I.; Schafer, A.J.; Harley, V.R. Compound effects of point mutations causing campomelic dysplasia/autosomal sex reversal upon SOX9 structure, nuclear transport, DNA binding, and transcriptional activation. J. Biol. Chem. 2001, 276, 27864–27872. [Google Scholar] [CrossRef] [PubMed]
- McDowall, S.; Argentaro, A.; Ranganathan, S.; Weller, P.; Mertin, S.; Mansour, S.; Tolmie, J.; Harley, V. Functional and structural studies of wild type SOX9 and mutations causing campomelic dysplasia. J. Biol. Chem. 1999, 274, 24023–24030. [Google Scholar] [CrossRef] [PubMed]
- Poulat, F.; Soullier, S.; Gozé, C.; Heitz, F.; Calas, B.; Berta, P. Description and functional implications of a novel mutation in the sex-determining gene SRY. Hum. Mutat. 1994, 3, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.K.; Jain, M.; Natarajan, S.; Frasier, S.D.; Vilain, E. Familial mutation in the testis-determining gene SRY shared by an XY female and her normal father. J. Clin. Endocrinol. Metab. 2002, 87, 3428–3432. [Google Scholar] [CrossRef]
- Thong, M.K.; Scherer, G.; Kozlowski, K.; Haan, E.; Morris, L. Acampomelic campomelic dysplasia with SOX9 mutation. Am. J. Med. Genet. 2000, 93, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Feng, C.-W.A.; Spiller, C.; Merriner, D.J.; O’Bryan, M.K.; Bowles, J.; Koopman, P. SOX30 is required for male fertility in mice. Sci. Rep. 2017, 7, 17619. [Google Scholar] [CrossRef]
- Béranger, F.; Méjean, C.; Moniot, B.; Berta, P.; Vandromme, M. Muscle differentiation is antagonized by SOX15, a new member of the SOX protein family. J. Biol. Chem. 2000, 275, 16103–16109. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Göring, W.; Ochs, M.; Mühlfeld, C.; Steding, G.; Paprotta, I.; Engel, W.; Adham, I.M. Sox15 is required for skeletal muscle regeneration. Mol. Cell. Biol. 2004, 24, 8428–8436. [Google Scholar] [CrossRef] [PubMed]
- Merchan-Sala, P.; Nardini, D.; Waclaw, R.R.; Campbell, K. Selective neuronal expression of the SoxE factor, Sox8, in direct pathway striatal projection neurons of the developing mouse brain. J. Comp. Neurol. 2017, 525, 2805–2819. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Lee, C.J.; Badge, R.M.; Orme, A.T.; Scotting, P.J. Sox8 gene expression identifies immature glial cells in developing cerebellum and cerebellar tumours. Brain Res. Mol. Brain Res. 2001, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, I.; Fernández-Alvarez, P.; Plaja, A.; Ariceta, G.; Sabaté-Rotés, A.; García-Arumí, E.; Vendrell, T.; Tizzano, E. Further delineation of the SOX18-related Hypotrichosis, Lymphedema, Telangiectasia syndrome (HTLS). Eur. J. Med. Genet. 2018, 61, 269–272. [Google Scholar] [CrossRef]
- Downes, M.; François, M.; Ferguson, C.; Parton, R.G.; Koopman, P. Vascular defects in a mouse model of hypotrichosis-lymphedema-telangiectasia syndrome indicate a role for SOX18 in blood vessel maturation. Hum. Mol. Genet. 2009, 18, 2839–2850. [Google Scholar] [CrossRef] [PubMed]
- Fontijn, R.D.; Volger, O.L.; Fledderus, J.O.; Reijerkerk, A.; de Vries, H.E.; Horrevoets, A.J.G. SOX-18 controls endothelial-specific claudin-5 gene expression and barrier function. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H891–H900. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Greene, C.; Hanley, N.; Reschke, C.R.; Reddy, A.; Mäe, M.A.; Connolly, R.; Behan, C.; O’Keeffe, E.; Bolger, I.; Hudson, N.; et al. Microvascular stabilization via blood-brain barrier regulation prevents seizure activity. Nat. Commun. 2022, 13, 2003. [Google Scholar] [CrossRef]
- Dailey, C.; Oshodi, R.B.; Boull, C.; Aggarwal, A. Expanding the clinical spectrum of SOX18-related Hypotrichosis-lymphedema-telangiectasia-renal defect syndrome. Eur. J. Med. Genet. 2022, 65, 104607. [Google Scholar] [CrossRef]
- Nowakowska, B.A.; Obersztyn, E.; Szymańska, K.; Bekiesińska-Figatowska, M.; Xia, Z.; Ricks, C.B.; Bocian, E.; Stockton, D.W.; Szczałuba, K.; Nawara, M.; et al. Severe mental retardation, seizures, and hypotonia due to deletions of MEF2C. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1042–1051. [Google Scholar] [CrossRef]
- Vrečar, I.; Innes, J.; Jones, E.A.; Kingston, H.; Reardon, W.; Kerr, B.; Clayton-Smith, J.; Douzgou, S. Further Clinical Delineation of the MEF2C Haploinsufficiency Syndrome: Report on New Cases and Literature Review of Severe Neurodevelopmental Disorders Presenting with Seizures, Absent Speech, and Involuntary Movements. J. Pediatr. Genet. 2017, 6, 129–141. [Google Scholar] [CrossRef]
- Borlot, F.; Whitney, R.; Cohn, R.D.; Weiss, S.K. MEF2C-related epilepsy: Delineating the phenotypic spectrum from a novel mutation and literature review. Seizure 2019, 67, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Saitsu, H.; Endo, W.; Kikuchi, A.; Uematsu, M.; Haginoya, K.; Hino-fukuyo, N.; Kobayashi, T.; Iwasaki, M.; Tominaga, T.; et al. RBPJ is disrupted in a case of proximal 4p deletion syndrome with epilepsy. Brain Dev. 2014, 36, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Tang, P.; Liu, S.; Dewing, P.; Harley, V.R.; Vilain, E. Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum. Mol. Genet. 2003, 12, 1755–1765. [Google Scholar] [CrossRef]
- Overman, J.; Fontaine, F.; Moustaqil, M.; Mittal, D.; Sierecki, E.; Sacilotto, N.; Zuegg, J.; Robertson, A.A.; Holmes, K.; Salim, A.A.; et al. Pharmacological targeting of the transcription factor SOX18 delays breast cancer in mice. eLife 2017, 6, e21221. [Google Scholar] [CrossRef]
- Restrepo, N.A.; Cooke Bailey, J.N. Primary Open-Angle Glaucoma Genetics in African Americans. Curr. Genet. Med. Rep. 2017, 5, 167–174. [Google Scholar] [CrossRef]
- Zhou, Y.; Williams, J.; Smallwood, P.M.; Nathans, J. Sox7, Sox17, and Sox18 Cooperatively Regulate Vascular Development in the Mouse Retina. PloS ONE 2015, 10, e0143650. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Gonen, N.; Sinclair, A. SOX Genes and Their Role in Disorders of Sex Development. Sex. Dev. 2022, 16, 80–91. [Google Scholar] [CrossRef]
- Prokop, J.W.; Deschepper, C.F. Chromosome Y genetic variants: Impact in animal models and on human disease. Physiol. Genom. 2015, 47, 525–537. [Google Scholar] [CrossRef]
- Kemena, C.; Notredame, C. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 2009, 25, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Brookes, A.J.; Robinson, P.N. Human genotype-phenotype databases: Aims, challenges and opportunities. Nat. Rev. Genet. 2015, 16, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Merino, F.; Ng, C.K.L.; Veerapandian, V.; Schöler, H.R.; Jauch, R.; Cojocaru, V. Structural basis for the SOX-dependent genomic redistribution of OCT4 in stem cell differentiation. Structure 2014, 22, 1274–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.E.; Kuchenbaecker, K.; Walters, R.K.; Chen, C.-Y.; Popejoy, A.B.; Periyasamy, S.; Lam, M.; Iyegbe, C.; Strawbridge, R.J.; Brick, L.; et al. Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell 2019, 179, 589–603. [Google Scholar] [CrossRef]
- The All of Us Research Program Investigators; Denny, J.C.; Rutter, J.L.; Goldstein, D.B.; Philippakis, A.; Smoller, J.W.; Jenkins, G.; Dishman, E. The “All of Us” Research Program. N. Engl. J. Med. 2019, 381, 668–676. [Google Scholar] [CrossRef]







| UniProt | Bowles/Koopman | AA Used | AA Used | AA Conservation | Publications | Unique ClinVar | gnomAD Count | HPO Profiles | Variant | Syn (s) | Nonsyn (n) | Conservation Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −1 | 2 | 3 | RHK | 0.93 ± 0.08 | 2 | 6 | 1 | SOX4 H58P | 4 | 0 | 1 | |
| 1 | 3 | 2 | VI | 0.92 ± 0.09 | 3 | 3 | 6 | 3 | SOX4 I59S | 7 | 0 | 1.25 |
| SOX9 V105F | 11 | 0 | 1 | |||||||||
| 2 | 4 | 2 | KR | 0.94 ± 0.07 | 3 | 3 | 1 | SOX3 K140R | 10 | 0 | 2 | |
| SOX9 K106E | 9 | 0 | 1.25 | |||||||||
| SOX10 K105Q | 4 | 0 | 1 | |||||||||
| 3 | 5 | 1 | R | 0.94 ± 0.07 | 2 | 10 | 2 | 0 | SOX4 R61Q | 22 | 0 | 1.5 |
| SOX5 R558H | 10 | 0 | 1.25 | |||||||||
| SOX6 R623Q | 9 | 0 | 1.25 | |||||||||
| SOX10 R106G | 15 | 0 | 1.25 | |||||||||
| 5 | 7 | 1 | M | 0.95 ± 0.05 | 2 | 4 | 21 | 2 | SOX6 M625T | 0 | 0 | 1 |
| SOX10 M108T | 0 | 0 | 1 | |||||||||
| 6 | 8 | 1 | N | 0.95 ± 0.05 | 3 | 1152 | 1 | SOX5 N561H | 10 | 0 | 1.5 | |
| SOX10 N109S | 16 | 0 | 2 | |||||||||
| 7 | 9 | 1 | A | 0.95 ± 0.05 | 4 | 2 | 0 | SRY A66P | 6 | 0 | 1.5 | |
| SOX4 A65T | 17 | 0 | 1.25 | |||||||||
| SOX9 A111T | 13 | 0 | 1 | |||||||||
| SOX10 A110V | 2 | 0 | 1 | |||||||||
| 9 | 11 | 2 | MI | 0.95 ± 0.04 | 4 | 9 | 4 | 0 | SOX9 M113V/I | 0 | 0 | 1 |
| SOX10 M112V/R/T/I | 0 | 0 | 1 | |||||||||
| 10 | 12 | 1 | V | 0.95 ± 0.04 | 1 | 5 | 0 | SOX9 V114L | 12 | 0 | 1 | |
| 11 | 13 | 1 | W | 0.95 ± 0.06 | 1 | 4 | 1 | 0 | SOX11 W59R | 0 | 0 | 1 |
| SOX9 W115R | 0 | 0 | 1 | |||||||||
| SOX6 W631C | 0 | 0 | 1 | |||||||||
| 12 | 14 | 2 | SA | 0.96 ± 0.03 | 5 | 2 | 0 | SOX11 S60P | 20 | 0 | 1.5 | |
| SOX9 A116V | 23 | 0 | 1.5 | |||||||||
| 15 | 17 | 4 | QEAH | 0.95 ± 0.04 | 1 | 1 | 29 | 2 | SOX9 A119E | 15 | 0 | 1.25 |
| 16 | 18 | 1 | R | 0.95 ± 0.04 | 10 | 4 | 0 | SOX2 R56G/W | 13 | 0 | 1.25 | |
| SOX9 R120G/L | 20 | 0 | 1.25 | |||||||||
| SOX11 R64C/G/H | 16 | 0 | 1.25 | |||||||||
| SOX10 R119L | 8 | 0 | 1 | |||||||||
| SOX5 R571L/W | 16 | 0 | 1.25 | |||||||||
| 19 | 21 | 3 | MIL | 0.93 ± 0.08 | 1 | 2 | 6 | 0 | SOX4 I77V | 12 | 0 | 1.5 |
| SOX10 L122V | 20 | 0 | 1.25 | |||||||||
| 20 | 22 | 3 | AML | 0.94 ± 0.09 | 1 | 4 | 9 | 0 | SOX9 A124P | 35 | 1 | 1 |
| SOX10 A123P | 15 | 0 | 1.25 | |||||||||
| 24 | 26 | 1 | P | 0.94 ± 0.10 | 1 | 14 | 6 | SRY P83H | 2 | 0 | 1.25 | |
| 26 | 28 | 3 | MLA | 0.94 ± 0.09 | 3 | 10 | 2 | SOX10 L129P | 11 | 0 | 1 | |
| 28 | 30 | 1 | N | 0.94 ± 0.09 | 2 | 3 | 16 | 0 | SOX11 N76D | 4 | 0 | 1 |
| SOX5 N583S | 9 | 0 | 1.25 | |||||||||
| SOX17 N95S | 9 | 0 | 1.5 | |||||||||
| 29 | 31 | 2 | SA | 0.94 ± 0.08 | 2 | 3 | 0 | SOX10 A132G/V | 19 | 0 | 1.5 | |
| 31 | 33 | 2 | IL | 0.94 ± 0.10 | 3 | 3 | 4 | 2 | SOX11 I79L | 12 | 0 | 1.5 |
| SOX10 L134P | 4 | 0 | 1 | |||||||||
| 32 | 34 | 1 | S | 0.94 ± 0.11 | 1 | 7 | 3 | 0 | SOX11 S80F/C | 2 | 0 | 1 |
| SOX5 S587C | 1 | 0 | 1 | |||||||||
| SOX10 S135R/G/T | 5 | 0 | 1 | |||||||||
| 33 | 35 | 2 | KV | 0.93 ± 0.10 | 1 | 1 | 0 | 0 | SOX5 K588N | 5 | 0 | 1 |
| 34 | 36 | 5 | RQITM | 0.95 ± 0.06 | 2 | 21 | 9 | SOX2 R74P | 16 | 0 | 1.5 | |
| SOX9 T138K | 27 | 0 | 1.5 | |||||||||
| 35 | 37 | 1 | L | 0.95 ± 0.07 | 1 | 1 | 1 | 1 | SOX4 L93Q | 11 | 0 | 1 |
| 36 | 38 | 1 | G | 0.95 ± 0.07 | 2 | 6 | 3 | 2 | SOX2 G76D | 8 | 0 | 1.25 |
| SRY G95R/E | 2 | 0 | 1.25 | |||||||||
| SOX9 G140D | 14 | 0 | 1 | |||||||||
| SOX10 G139C/D | 10 | 0 | 1 | |||||||||
| 38 | 40 | 7 | EDQRLSA | 0.93 ± 0.10 | 2 | 23 | 3 | SOX10 L141P | 11 | 0 | 1 | |
| 39 | 41 | 1 | W | 0.96 ± 0.06 | 1 | 7 | 1 | 0 | SOX4 W97G | 0 | 0 | 1 |
| SOX6 W659R | 0 | 0 | 1 | |||||||||
| SOX10 W142R/S/C | 0 | 0 | 1 | |||||||||
| 47 | 49 | 1 | K | 0.96 ± 0.06 | 2 | 3 | 24 | 0 | SRY K106I | 1 | 0 | 1.25 |
| SOX10 K150E | 5 | 0 | 1 | |||||||||
| SOX4 K105N | 1 | 0 | 1 | |||||||||
| 48 | 50 | 5 | RWIQK | 0.96 ± 0.05 | 1 | 2 | 12 | 0 | SOX10 R151P | 15 | 0 | 1.25 |
| 50 | 52 | 2 | FY | 0.96 ± 0.02 | 2 | 5 | 11 | 0 | SRY F109S | 2 | 0 | 1.25 |
| SOX9 F154L | 14 | 0 | 1.5 | |||||||||
| SOX10 F153I | 13 | 0 | 1.5 | |||||||||
| 52 | 54 | 4 | DQRE | 0.92 ± 0.13 | 2 | 11 | 1 | SOX10 E155K | 6 | 0 | 1 | |
| 53 | 55 | 1 | E | 0.97 ± 0.02 | 1 | 285 | 16 | SOX18 E137K | 10 | 0 | 2 | |
| 54 | 56 | 2 | AQ | 0.97 ± 0.03 | 3 | 6 | 9 | 1 | SRY A113T | 5 | 0 | 1.5 |
| SOX11 A102V | 16 | 0 | 1.25 | |||||||||
| SOX10 A157V | 28 | 0 | 2 | |||||||||
| SOX4 A112P | 11 | 0 | 1.25 | |||||||||
| 55 | 57 | 4 | KQEA | 0.96 ± 0.04 | 2 | 9 | 1 | SOX17 E122D | 3 | 0 | 1 | |
| 56 | 58 | 2 | RK | 0.96 ± 0.07 | 4 | 14 | 4 | SOX2 R96P | 6 | 0 | 1 | |
| SOX9 R160P | 19 | 0 | 1.25 | |||||||||
| SOX5 R611G | 22 | 0 | 1.5 | |||||||||
| SOX6 R676Q | 13 | 0 | 1.25 | |||||||||
| 57 | 59 | 2 | LI | 0.97 ± 0.01 | 2 | 0 | 0 | SOX2 L97P | 5 | 0 | 1 | |
| SOX10 L160P | 12 | 0 | 1 | |||||||||
| 58 | 60 | 4 | RQSK | 0.97 ± 0.02 | 1 | 3 | 76 | 2 | SOX10 R161C/H | 15 | 0 | 1.25 |
| SOX17 R125S | 18 | 0 | 1.25 | |||||||||
| 61 | 63 | 1 | H | 0.95 ± 0.10 | 2 | 4 | 3 | 0 | SOX2 H101R | 1 | 0 | 1 |
| SOX9 H165Y/R | 7 | 0 | 1.25 | |||||||||
| SOX10 H164P | 5 | 0 | 1 | |||||||||
| 63 | 65 | 5 | KEAQR | 0.94 ± 0.10 | 1 | 26 | 2 | SOX10 K166E | 11 | 0 | 1.25 | |
| 65 | 67 | 3 | HYF | 0.95 ± 0.06 | 2 | 4 | 3 | SOX11 Y113C | 16 | 1 | 1 | |
| 68 | 70 | 2 | YW | 0.95 ± 0.08 | 3 | 6 | 4 | 0 | SOX11 Y116C | 3 | 0 | 1 |
| SOX17 Y135C | 3 | 0 | 1 | |||||||||
| 70 | 72 | 1 | Y | 0.95 ± 0.11 | 4 | 0 | 0 | SOX4 Y128H | 2 | 0 | 1 | |
| 71 | 73 | 3 | RKQ | 0.95 ± 0.08 | 1 | 3 | 11 | 4 | SOX10 Q174P | 8 | 0 | 1.25 |
| 72 | 74 | 1 | P | 0.93 ± 0.13 | 4 | 9 | 4 | 0 | SOX2 P112T/A | 11 | 0 | 1.25 |
| SOX11 P120L | 13 | 0 | 1.25 | |||||||||
| SOX9 P176T/S/L/R | 35 | 1 | 1 | |||||||||
| SOX6 P692S | 11 | 0 | 1.25 | |||||||||
| SOX10 P175S | 17 | 0 | 1.25 | |||||||||
| 73 | 75 | 1 | R | 0.91 ± 0.14 | 3 | 25 | 4 | SOX2 R113W | 4 | 0 | 1 | |
| 74 | 76 | 3 | RKP | 0.92 ± 0.13 | 1 | 3 | 6 | 0 | SOX10 R177Q | 25 | 0 | 1.5 |
| Gene | Geno2MP Var | HMG Box # | AA Used in Human | AA Conservation | Genes with ClinVar | Phenotype | Geno2MP HPO | Geno2MP CADD | gnomAD Count | Syn (s) | Nonsyn (n) | Conservation Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SOX1 | E88A | 38 | 7 | 0.93 ± 0.10 | 2 | Microcephaly | 1 | 21.6 | 4 | 5 | 0 | 1.5 |
| SOX10 | H128Q | 25 | 5 | 0.93 ± 0.10 | 1 | Abnormality of the ear | 1 | 23.8 | 0 | 4 | 0 | 1 |
| SOX10 | L138P | 35 | 1 | 0.95 ± 0.07 | 1 | Nephrotic syndrome | 1 | 21.2 | 0 | 11 | 0 | 1 |
| SOX11 | G84S | 36 | 1 | 0.95 ± 0.07 | 4 | Abnormality of the globe | 1 | 28.5 | 0 | 13 | 0 | 1.25 |
| SOX13 | Q444E | 21 | 6 | 0.92 ± 0.09 | 0 | Abnormality of the limb | 2 | 28.6 | 1 | 16.5 | 2.5 | 1 |
| SOX13 | R461H | 38 | 7 | 0.93 ± 0.10 | 2 | Hypoplastic left-heart | 2 | 34 | 8 | 17 | 0 | 1.25 |
| SOX13 | A478E | 55 | 4 | 0.96 ± 0.04 | 2 | Retinitis pigmentosa | 1 | 28.5 | 2 | 24 | 0 | 1.5 |
| SOX14 | R63Q | 56 | 2 | 0.96 ± 0.07 | 4 | Congenital diaphragmatic hernia | 2 | 29.2 | 1 | 19 | 1 | 1 |
| SOX14 | R80Q | 73 | 1 | 0.91 ± 0.14 | 2 | Abnormal muscle physiology (×2) | 3 | 26 | 0 | 11 | 0 | 1.25 |
| SOX15 | R82L | 34 | 5 | 0.95 ± 0.06 | 2 | Retinitis pigmentosa | 2 | 36 | 9 | 7 | 0 | 1.25 |
| SOX15 | G84D | 36 | 1 | 0.95 ± 0.07 | 4 | Tricuspid atresia | 1 | 33 | 2 | 11 | 0 | 1.25 |
| SOX15 | R104G | 56 | 2 | 0.96 ± 0.07 | 4 | Progressive muscle weakness | 2 | 27.2 | 0 | 14 | 0 | 1.25 |
| SOX17 | M101V | 34 | 5 | 0.95 ± 0.06 | 2 | Distal arthrogryposis | 1 | 23.2 | 0 | 0 | 0 | 1 |
| SOX17 | R125S | 58 | 4 | 0.97 ± 0.02 | 2 | Neural Atrophy/Degeneration | 1 | 33 | 4 | 18 | 0 | 1.25 |
| SOX18 | E137K | 53 | 1 | 0.97 ± 0.02 | 1 | Seizures (×2), DD/ID, ASD, brain morphology, heart | 16 | 28.6 | 278 | 10 | 0 | 2 |
| SOX18 | N151S | 67 | 3 | 0.95 ± 0.05 | 0 | abdominal organs | 1 | 24 | 1 | 3 | 0 | 1 |
| SOX18 | R155Q | 71 | 3 | 0.95 ± 0.08 | 2 | Malformation of the heart (×2) | 3 | 26.6 | 1 | 15 | 0 | 1.25 |
| SOX30 | I367V | 31 | 2 | 0.94 ± 0.10 | 3 | ASD, Cerebellar hypoplasia (×2) | 2 | 22.5 | 0 | 3 | 0 | 1 |
| SOX30 | E399A | 63 | 5 | 0.94 ± 0.10 | 1 | cardiovascular system | 1 | 24.5 | 3 | 7 | 0 | 1.5 |
| SOX6 | D607E | 14 | 5 | 0.91 ± 0.13 | 0 | Abnormality of the cerebral cortex | 1 | 25.1 | 0 | 16 | 0 | 2 |
| SOX6 | M619T | 26 | 3 | 0.94 ± 0.09 | 3 | Thoracic aortic aneurysm | 1 | 21.5 | 0 | 0 | 0 | 1 |
| SOX7 | K81R | 37 | 6 | 0.92 ± 0.12 | 0 | Spontaneous abortion | 2 | 22.2 | 1 | 12 | 0 | 1.25 |
| SOX7 | A98G | 54 | 2 | 0.97 ± 0.03 | 5 | Abnormality of hindbrain morphology | 1 | 27.8 | 1 | 33 | 0 | 1.5 |
| SOX7 | D108N | 64 | 3 | 0.95 ± 0.11 | 2 | central nervous system | 2 | 35 | 3 | 17 | 0 | 1.5 |
| SOX8 | R159G | 58 | 4 | 0.97 ± 0.02 | 2 | Intellectual disability, microcephaly (×2) | 1 | 20.8 | 1 | 40 | 0 | 1.5 |
| SOX8 | K163R | 62 | 4 | 0.96 ± 0.02 | 0 | Nephrotic syndrome | 1 | 35 | 5 | 10 | 0 | 1.25 |
| Gene | Codon # | AA | Var | Phenotype | Syn (s) | Nonsyn (n) | Selection Score | 21 Codon Window | AA Conservation | gnomAD Count | Geno2MP | HPO Profiles | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SOX1 | 126 | T | T126I | Abnormality of hindbrain morphology | 1 | 0 | 1 | 25.25 | 0.97 | 1 | T126I | 1 | 21.7 |
| SOX2 | 123 | D | D123G | Developmental disorder | 4 | 0 | 1.25 | 17.5 | 1.00 | 1 | |||
| SOX2 | 130 | G | G130A | abnormalities of the central nervous system | 8 | 0 | 1.25 | 15.25 | 0.99 | 17 | G130A | 1 | 23.3 |
| SOX2 | 133 | A | A133T | Anophthalmia/microphthalmia-esophageal atresia syndrome | 22 | 0 | 2 | 11.25 | 0.96 | 8 | A133T | 3 | 23.3 |
| SOX2 | 272 | D | D272N | not provided | 1 | 0 | 1 | 13.75 | 0.97 | ||||
| SOX5 | 135 | R | R122H | Intellectual disability | 10 | 0 | 1.25 | 23.75 | 0.94 | 1 | R122H | 1 | 35 |
| SOX5 | 159 | P | P146L | Thoracic aortic aneurysm | 10 | 0 | 1.25 | 14.75 | 0.94 | 1 | P146L | 1 | 25.1 |
| SOX5 | 206 | I | I206V | Lamb-Shaffer syndrome | 4 | 0 | 1 | 19.75 | 0.95 | ||||
| SOX5 | 228 | A | A215V | Coarctation of aorta | 22 | 0 | 1.5 | 20.5 | 0.95 | 10 | A215V | 1 | 33 |
| SOX5 | 261 | K | K261N | not provided | 4 | 0 | 1 | 22.5 | 0.96 | ||||
| SOX5 | 266 | Q | Q266H | Lamb-Shaffer syndrome | 7 | 0 | 1.25 | 16.25 | 0.96 | ||||
| SOX5 | 268 | Q | Q268H | Lamb-Shaffer syndrome | 5 | 0 | 1 | 14.5 | 0.96 | ||||
| SOX6 | 146 | E | E146K | not provided | 10 | 0 | 1.5 | 13 | 0.95 | 2 | E146D | 2 | 20.4 |
| SOX6 | 209 | H | H209K | Multiple neurological | 16 | 0 | 2 | 22.25 | 0.97 | ||||
| SOX6 | 214 | K | K214Q | Hypoplastic left-heart syndrome | 8 | 0 | 1.25 | 23.5 | 0.97 | 4 | K214Q | 2 | 22 |
| SOX6 | 252 | N | N252K | not provided | 3 | 0 | 1 | 18.25 | 0.97 | ||||
| SOX6 | 264 | M | M264I | myopathy | 0 | 0 | 1 | 17.25 | 0.96 | 5 | M264I | 1 | 22.1 |
| SOX6 | 277 | R | R277W | not provided | 7 | 0 | 1 | 19.75 | 0.97 | 14 | |||
| SOX6 | 280 | A | A280E | Aplasia/Hypoplasia affecting the eye | 4 | 0 | 1 | 18.75 | 0.97 | 1 | A280E | 2 | 23.1 |
| SOX6 | 281 | A | A281T | not provided | 8 | 0 | 1.25 | 19 | 0.97 | 1 | |||
| SOX6 | 291 | F | F291L | cardiovascular system | 7 | 0 | 1.25 | 17 | 0.97 | 5 | F291L | 1 | 22.4 |
| SOX6 | 310 | S | S310T | Microcephaly | 5 | 0 | 1 | 13.25 | 0.97 | 56 | S310T | 2 | 21.6 |
| SOX6 | 312 | M | M312V | not provided | 0 | 0 | 1 | 13.75 | 0.95 | ||||
| SOX6 | 371 | A | A330P | Aplasia/Hypoplasia affecting the eye | 10 | 0 | 1.25 | 14 | 0.99 | 1 | A330P | 1 | 23.9 |
| SOX6 | 512 | R | R485Q | Abnormality of nervous system | 12 | 0 | 1.25 | 17 | 0.99 | 2 | R485Q | 1 | 28.5 |
| SOX6 | 572 | R | R545Q | Intellectual disability | 9 | 0 | 1.25 | 15 | 0.99 | 6 | R545Q | 1 | 32 |
| SOX6 | 618 | E | E591K | Intellectual disability | 10 | 0 | 1.5 | 20.25 | 0.98 | 1 | E591K | 1 | 35 |
| SOX7 | 128 | R | R128C | abnormality of the central nervous system | 30 | 0 | 1.5 | 12 | 0.99 | 4 | R128C | 1 | 21.8 |
| SOX7 | 329 | R | R329H | Nephrotic syndrome | 19 | 0 | 1.25 | 15.25 | 1.00 | 3 | R329H | 1 | 25.3 |
| SOX7 | 379 | A | A379V | Abnormality of the eye (×4) | 25 | 0 | 1.25 | 21 | 0.99 | 195 | A379V | 9 | 29 |
| SOX8 | 263 | N | N263I | Abnormality of the nervous system (×2) | 17 | 0 | 1.25 | 13.25 | 0.99 | 45 | N263I | 6 | 24.1 |
| SOX9 | 73 | I | I73T | Campomelic dysplasia | 2 | 0 | 1 | 14.25 | 0.97 | ||||
| SOX9 | 76 | A | A76E | Inborn genetic diseases | 32 | 0 | 2 | 17 | 0.97 | ||||
| SOX9 | 81 | L | L81V | Campomelic dysplasia | 20 | 0 | 1.25 | 18.25 | 0.97 | 1 | |||
| SOX9 | 83 | G | G83R | Inborn genetic diseases | 10 | 0 | 1 | 20.25 | 0.97 | ||||
| SOX10 | 68 | F | F68L | PCWH syndrome | 4 | 0 | 1 | 12.5 | 0.96 | 1 | |||
| SOX10 | 75 | A | A75V | Malformation of the heart and great vessels (×2) | 20 | 0 | 1.5 | 17.25 | 0.96 | A75V | 2 | 34 | |
| SOX10 | 92 | V | V92M/L | PCWH syndrome | 7 | 0 | 1 | 17.5 | 0.97 | 46 | V92L | 4 | 24.1 |
| SOX10 | 179 | K | K179N | not provided | 3 | 0 | 1 | 19.25 | 1.00 | ||||
| SOX10 | 181 | G | G181R | not provided | 9 | 0 | 1 | 17.5 | 0.98 | 1 | |||
| SOX10 | 216 | H | H216Q | not provided | 4 | 0 | 1 | 13 | 0.74 | ||||
| SOX10 | 240 | T | T240P | Waardenburg syndrome type 4C | 12 | 0 | 1.25 | 16.25 | 1.00 | ||||
| SOX10 | 278 | I | I278V | Aganglionic megacolon | 15 | 0 | 1.5 | 12.75 | 1.00 | 25 | |||
| SOX10 | 428 | M | M428I | Abnormality of limb bone | 0 | 0 | 1 | 15.75 | 0.99 | 14 | M428I | 2 | 22.7 |
| SOX10 | 433 | R | R433Q | not provided | 6 | 0 | 1 | 19.5 | 0.99 | 2 | |||
| SOX11 | 417 | C | C417W | not provided | 4 | 0 | 1 | 17.25 | 0.95 | ||||
| SOX13 | 171 | S | S171L | Abnormality of the ear | 36 | 2 | 1 | 13 | 0.99 | 2 | S171L | 1 | 26.3 |
| SOX13 | 192 | R | R192Q | Multiple | 15 | 0 | 1.25 | 22.25 | 0.99 | 4 | R192Q | 5 | 34 |
| SOX13 | 210 | H | H210R | Neural Atrophy/Degeneration | 13 | 0 | 1.5 | 21.25 | 0.98 | 4 | H210R | 4 | 26.7 |
| SOX13 | 507 | R | R507Q | Muscular dystrophy | 16 | 0 | 1.25 | 19.5 | 0.96 | 3 | R507Q | 2 | 34 |
| SOX14 | 88 | K | K88R | Abnormality of the cardiovascular system (×9) | 4 | 0 | 1 | 23 | 0.95 | 52 | K88R | 8 | 25.8 |
| SOX14 | 187 | T | T187K | Hypoplastic left-heart syndrome | 5 | 0 | 1 | 21 | 1.00 | 1 | T187K | 2 | 25.4 |
| SOX18 | 326 | E | E326V | Skeletal muscle atrophy | 1 | 0 | 1 | 10.25 | 1.00 | E326V | 1 | 21.4 | |
| SOX18 | 331 | L | L331F | HLTS | 9 | 0 | 1 | 11 | 1.00 | ||||
| SOX18 | 369 | L | L369V | not provided | 19 | 0 | 1.25 | 16 | 1.00 | 1 | |||
| SOX18 | 375 | A | A375T | Nephrotic syndrome | 35 | 0 | 2 | 13.25 | 0.99 | A375T | 1 | 20.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Underwood, A.; Rasicci, D.T.; Hinds, D.; Mitchell, J.T.; Zieba, J.K.; Mills, J.; Arnold, N.E.; Cook, T.W.; Moustaqil, M.; Gambin, Y.; et al. Evolutionary Landscape of SOX Genes to Inform Genotype-to-Phenotype Relationships. Genes 2023, 14, 222. https://doi.org/10.3390/genes14010222
Underwood A, Rasicci DT, Hinds D, Mitchell JT, Zieba JK, Mills J, Arnold NE, Cook TW, Moustaqil M, Gambin Y, et al. Evolutionary Landscape of SOX Genes to Inform Genotype-to-Phenotype Relationships. Genes. 2023; 14(1):222. https://doi.org/10.3390/genes14010222
Chicago/Turabian StyleUnderwood, Adam, Daniel T Rasicci, David Hinds, Jackson T Mitchell, Jacob K Zieba, Joshua Mills, Nicholas E Arnold, Taylor W Cook, Mehdi Moustaqil, Yann Gambin, and et al. 2023. "Evolutionary Landscape of SOX Genes to Inform Genotype-to-Phenotype Relationships" Genes 14, no. 1: 222. https://doi.org/10.3390/genes14010222
APA StyleUnderwood, A., Rasicci, D. T., Hinds, D., Mitchell, J. T., Zieba, J. K., Mills, J., Arnold, N. E., Cook, T. W., Moustaqil, M., Gambin, Y., Sierecki, E., Fontaine, F., Vanderweele, S., Das, A. S., Cvammen, W., Sirpilla, O., Soehnlen, X., Bricker, K., Alokaili, M., ... Prokop, J. W. (2023). Evolutionary Landscape of SOX Genes to Inform Genotype-to-Phenotype Relationships. Genes, 14(1), 222. https://doi.org/10.3390/genes14010222