
SARS-CoV-2 Omicron Variant Genomic and Phylogenetic Analysis in Iraqi Kurdistan Region

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Consent to Participate
2.2. Sample Collection
2.3. RNA Isolation and RT-PCR for Omicron Identification
2.4. Data Analysis
2.5. Whole-Genome Sequencing, Genome Assembly and Phylogenetic Analysis
2.6. Bioinformatic Analysis
2.7. Phylogenetic Analysis
3. Results
3.1. Clinical Data of Patients Infected with Omicron
3.2. The Mutational Change in the Omicron Variant in the Kurdistan Region of Iraq
3.3. Quantitative Accumulation of Variations in the IKR Omicron Variant (B.1.1.529 + BA.1)
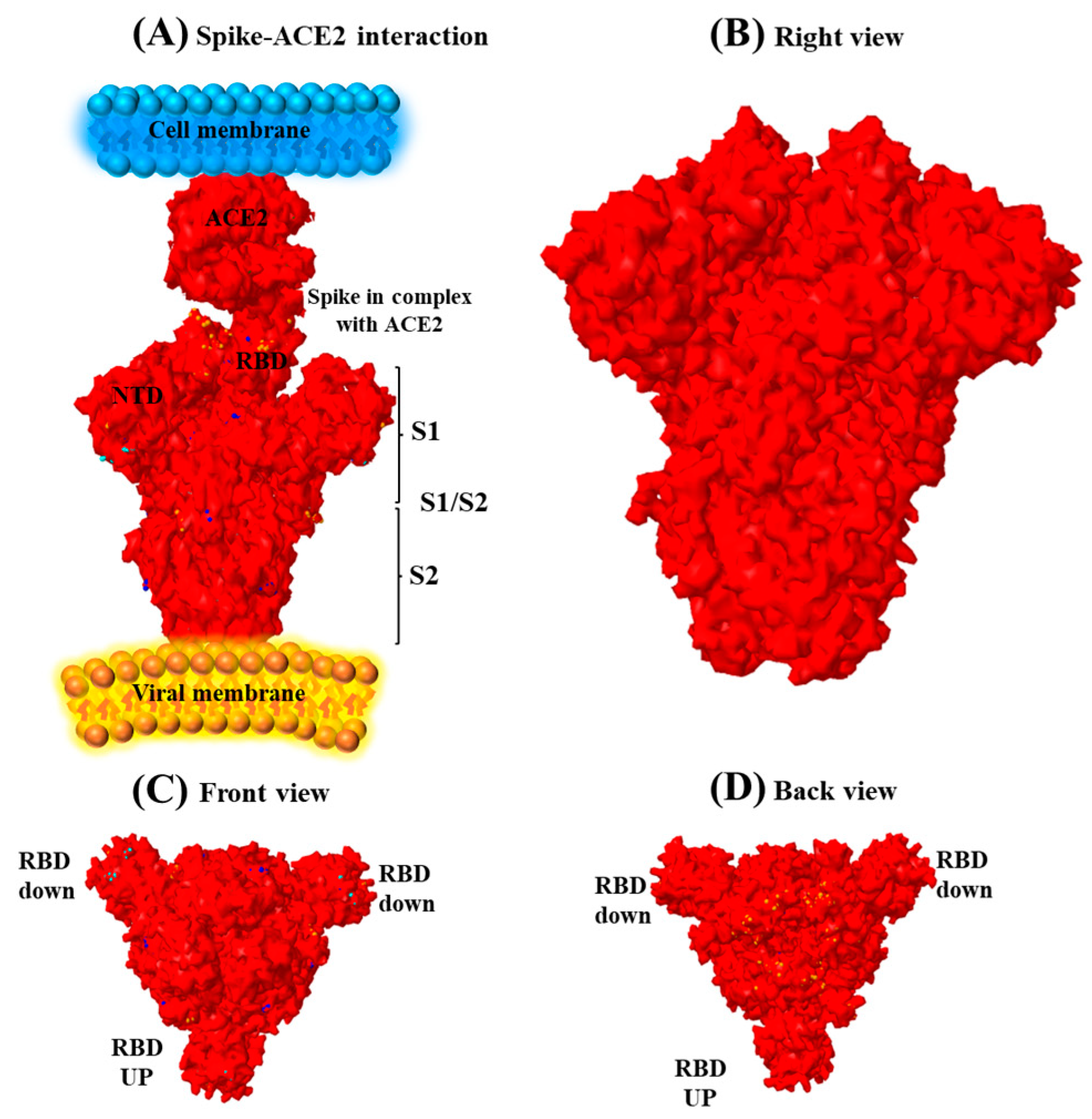
3.4. Spike Sequence Analysis of the Omicron Genome
3.5. Dominance of the Omicron Variant among the Kurdish Population during 2021
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. SPRP 2021 Mid-term Report-WHO Strategic Action Against COVID-19; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.T.; Perkins, J.S.; Mollett, G.; Blacow, R.M.; Consortium, C.-G.U.; Peacock, T.P.; et al. Reduced neutralisation of the Delta (B.1.617.2) SARS-CoV-2 variant of concern following vaccination. PLoS Pathog. 2021, 17, e1010022. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.; Bhattarai, N.; Hossen, M.L.; Stebliankin, V.; Gerstman, B.S.; Narasimhan, G.; Chapagain, P.P. Mutation-induced changes the receptor-binding interface of the SARS-CoV-2 Delta variant B. 1.617.2 and implications for immune evasion. Biochem. Biophys. Res. Commun. 2021, 574, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020. [Google Scholar] [CrossRef]
- Wruck, W.; Adjaye, J. Detailed phylogenetic analysis tracks transmission of distinct SARS-CoV-2 variants from China and Europe to West Africa. Sci. Rep. 2021, 11, 21108. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, J.P.; Samal, K.C. World on Alert: WHO Designated South African New COVID Strain (Omicron/B. 1.1. 529) as a Variant of Concern. Biot. Res. Today 2021, 3, 1086–1088. [Google Scholar]
- Keith, D. Clear Creek ISD Offering Virtual Learning Again as COVID-19 Cases Continue to Rise. FOX 26 Houston, 25 August 2021. [Google Scholar]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Insights on the mutational landscape of the SARS-CoV-2 Omicron variant receptor-binding domain. Cell Rep. Med. 2022, 3, 100527. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Sharma, A.R.; Dhama, K.; Agoramoorthy, G.; Chakraborty, C. Omicron variant (B.1.1.529) of SARS-CoV-2: Understanding mutations in the genome, S-glycoprotein, and antibody-binding regions. GeroScience 2022, 44, 619–637. [Google Scholar] [CrossRef]
- Song, Y.; Masaki, F. Preparation for the challenge of heavily mutated Omicron variant. Clin. Transl. Med. 2021, 11, e679. [Google Scholar] [CrossRef]
- Evans, J.P.; Zeng, C.; Qu, P.; Faraone, J.; Zheng, Y.M.; Carlin, C.; Bednash, J.S.; Zhou, T.; Lozanski, G.; Mallampalli, R.; et al. Neutralization of SARS-CoV-2 Omicron sub-lineages BA.1, BA.1.1, and BA.2. Cell Host Microbe 2022, 30, 1093–1102.e3. [Google Scholar] [CrossRef]
- He, C.; He, X.; Yang, J.; Lei, H.; Hong, W.; Song, X.; Yang, L.; Li, J.; Wang, W.; Shen, G.; et al. Spike protein of SARS-CoV-2 Omicron (B.1.1.529) variant have a reduced ability to induce the immune response. Signal Transduct. Target. Ther. 2022, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lin, X.; Chen, Z.; Yang, F.; Lin, S.; Yang, J.; Chen, H.; Sun, H.; Wang, L.; Wen, A.; et al. S19W, T27W, and N330Y mutations in ACE2 enhance SARS-CoV-2 S-RBD binding toward both wild-type and antibody-resistant viruses and its molecular basis. Signal Transduct. Target. Ther. 2021, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e2316. [Google Scholar] [CrossRef]
- Taboada, B.; Isa, P.; Gutiérrez-Escolano, A.; Del Ángel, R.; Ludert, J.; Vázquez, N.; Tapia-Palacios, M.; Chávez, P.; Garrido, E.; Espinosa, A. The geographic structure of viruses in the Cuatro Ciénegas Basin, a unique oasis in northern Mexico, reveals a highly diverse population on a small geographic scale. Appl. Environ. Microbiol. 2018, 84, e00418–e00465. [Google Scholar] [CrossRef]
- Illumina. TruSeq Stranded Total RNA Reference Guide; Illumina: San Diego, CA, USA, 2017. [Google Scholar]
- Andrews, J. Rugby Dads; Accent Press Ltd.: Pembroke Dock, UK, 2016. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Perez, J.C.; Lounnas, V.; Montagnier, M. The Omicron Variant Breaks the Evolutionary Lineage of Sars-Cov2 Variants. Int. J. Res.-Granthaalayah. 2021, 9, 108–132. [Google Scholar] [CrossRef]
- Wu, A.; Wang, L.; Zhou, H.Y.; Ji, C.Y.; Xia, S.Z.; Cao, Y.; Meng, J.; Ding, X.; Gold, S.; Jiang, T.; et al. One year of SARS-CoV-2 evolution. Cell Host Microbe 2021, 29, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, M.L.; Lei, Q.; Wang, F.; Hong, W.; Lai, D.Y.; Hou, H.; Xu, Z.W.; Zhang, B.; Chen, H.; et al. Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1051 COVID-19 patients. Cell Rep. 2021, 34, 108915. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Mohamed, M.E.M.; Abd El-Lateef, H.M.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2022, 94, 1627–1632. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Features | No. of Cases (%) | OR (95% C1) | p-Value for Interaction |
|---|---|---|---|
| Gender Male Female | 105 (60) 70 (40) | 8.21 (6.10–10.02) 6.03 (5.09–8.34) | 0.02 |
| Age 50< 50≥ | 110 (63) 65 (37) | 9.43 (7.06–12.3) 5.32 (4.21–6.89) | 0.01 |
| BMI Categories: Underweight ≤18.5 Normal weight = 18.5–24.9 Overweight = 25–29.9 Obesity = BMI of 30 or greater | 15 (9) 95 (54) 50 (30) 10 (7) | 4.20 (3.11–5.98) 8.89 (4.21–11.20) 5.32 (4.45–6.64) 4.01 (3.34–5.34) | 0.05 |
| Common Symptoms Fever Chills Cough Sore throat Runny nose Diarrhea | 120 (68.6) 149 (85.1) 129 (73.7) 97 (55.4) 127 (72.7) 170 (87.5) | 7.34 (5.11–9.21) 6.54 (5.67–8.43) 8.31 (6.98–10.22) 5.67 (6.34–7.84) 6.32 (5.32–7.89) 9.45 (8.34–11.45) | 0.02 0.21 0.42 0.31 0.23 |
| Vaccination Vaccinated Non-vaccinated | 65 (37) 110 (63) | 5.84 (4.32–6.45) 8.83 (6.38–10.89) | 0.01 |
| Hospitalized with Omicron Non-hospitalized Hospitalized | 125 (71) 50 (29) | 11.1 (9.43–12.34) 6.34 (4.50–7.69) | 0.05 |
| BA.1 Structure | List of Amino Acid Changes |
|---|---|
| NSP3 NSP4 NSP5 NSP6 NSP8 NSP12 NSP14 | K38R, S165del, L1266I, A1892T T492I P132H L105del, S106del, I189V, Q208R, C209del, I210del, R233del, Y234H, R252del, Y253del, L206P A86del, M87V F283S, P323LI47V |
Spike glycoprotein | Spike A67V, Spike T95I, Spike G142D, Spike L212I, Spike S375F, Spike T547K, Spike H655K, Spike N679K, Spike N764K, Spike N856K, Spike H954Q, Spike N969K, Spike L981F Spike H96del, Spike V70deI, Spike Y145del, ins214EPE, Spike N211del, Spike V143del Spike Y144del, Spike G339D, Spike S373P, Spike S375F, Spike S477N, Spike T478K, Spike E484A, Spike Q493R, Spike G496S, Spike Q498R, Spike N501Y, Spike Y505H, Spike D614G, Spike P681H, Spike D796Y |
| Envelope Membrane Nuclear | T9I D3G, Q19E, A63T P13L, E31del, R32del, S33del, R203K, G204R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majed, S.O.; Mustafa, S.A.; Jalal, P.J.; Fatah, M.H.; Miasko, M.; Jawhar, Z.; Karim, A.Y. SARS-CoV-2 Omicron Variant Genomic and Phylogenetic Analysis in Iraqi Kurdistan Region. Genes 2023, 14, 173. https://doi.org/10.3390/genes14010173
Majed SO, Mustafa SA, Jalal PJ, Fatah MH, Miasko M, Jawhar Z, Karim AY. SARS-CoV-2 Omicron Variant Genomic and Phylogenetic Analysis in Iraqi Kurdistan Region. Genes. 2023; 14(1):173. https://doi.org/10.3390/genes14010173
Chicago/Turabian StyleMajed, Sevan Omer, Suhad Asad Mustafa, Paywast Jamal Jalal, Mohammed Hassan Fatah, Monika Miasko, Zanko Jawhar, and Abdulkarim Yasin Karim. 2023. "SARS-CoV-2 Omicron Variant Genomic and Phylogenetic Analysis in Iraqi Kurdistan Region" Genes 14, no. 1: 173. https://doi.org/10.3390/genes14010173
APA StyleMajed, S. O., Mustafa, S. A., Jalal, P. J., Fatah, M. H., Miasko, M., Jawhar, Z., & Karim, A. Y. (2023). SARS-CoV-2 Omicron Variant Genomic and Phylogenetic Analysis in Iraqi Kurdistan Region. Genes, 14(1), 173. https://doi.org/10.3390/genes14010173