
Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs

 and
and 
Abstract
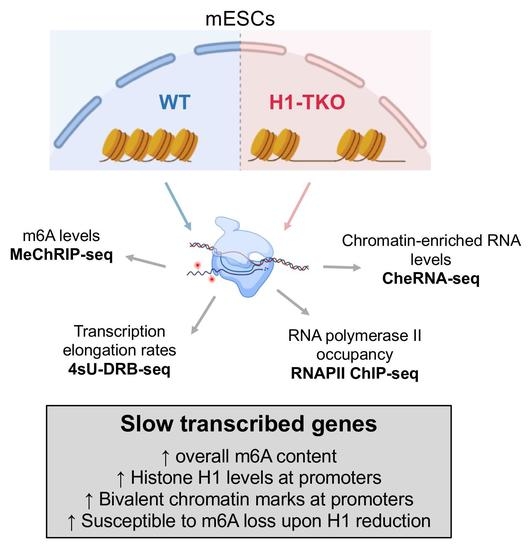
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. 4sU-DRB Sequencing (DRB-TT Seq)
2.3. Calculation of Transcription Elongation Rates
2.4. Mononucleosomal DNA Sequencing (MN-Seq) Analyses
2.5. Chromatin Enriched RNA (CheRNA) Sequencing Analyses
2.6. RNAPII Chromatin Immunoprecipitation (ChIP) Sequencing Analyses
2.7. m6A-CheRNA Immunoprecipitation (MeChRIP) Sequencing Analyses
2.8. Statistical Analyses
2.9. GO-Term Analysis
2.10. m6A Peak-Calling
2.11. Deposited Datasets
3. Results
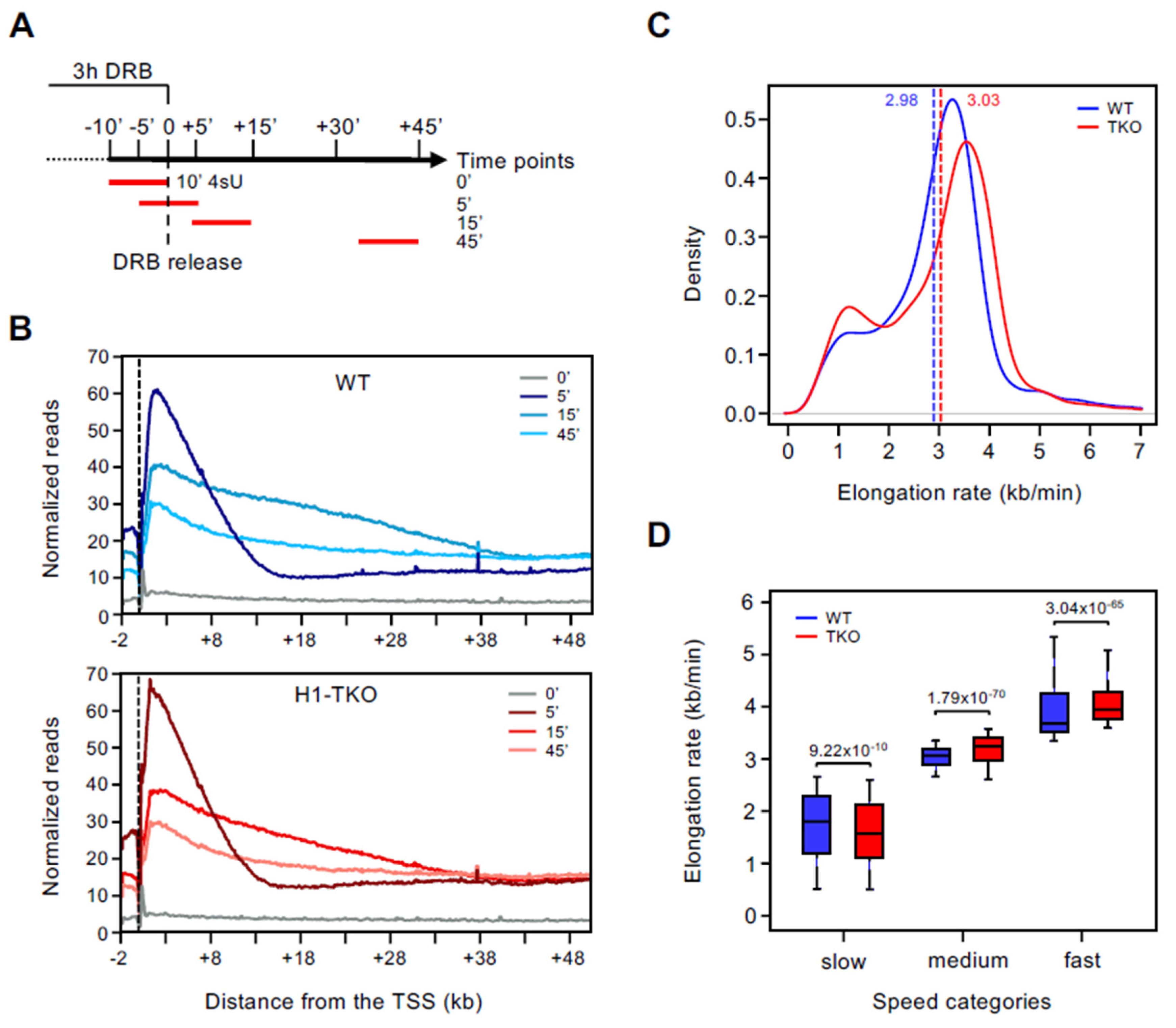
3.1. Reduction in Histone H1 Content Affects RNAPII Elongation Speeds at Slow and Fast Genes Differently
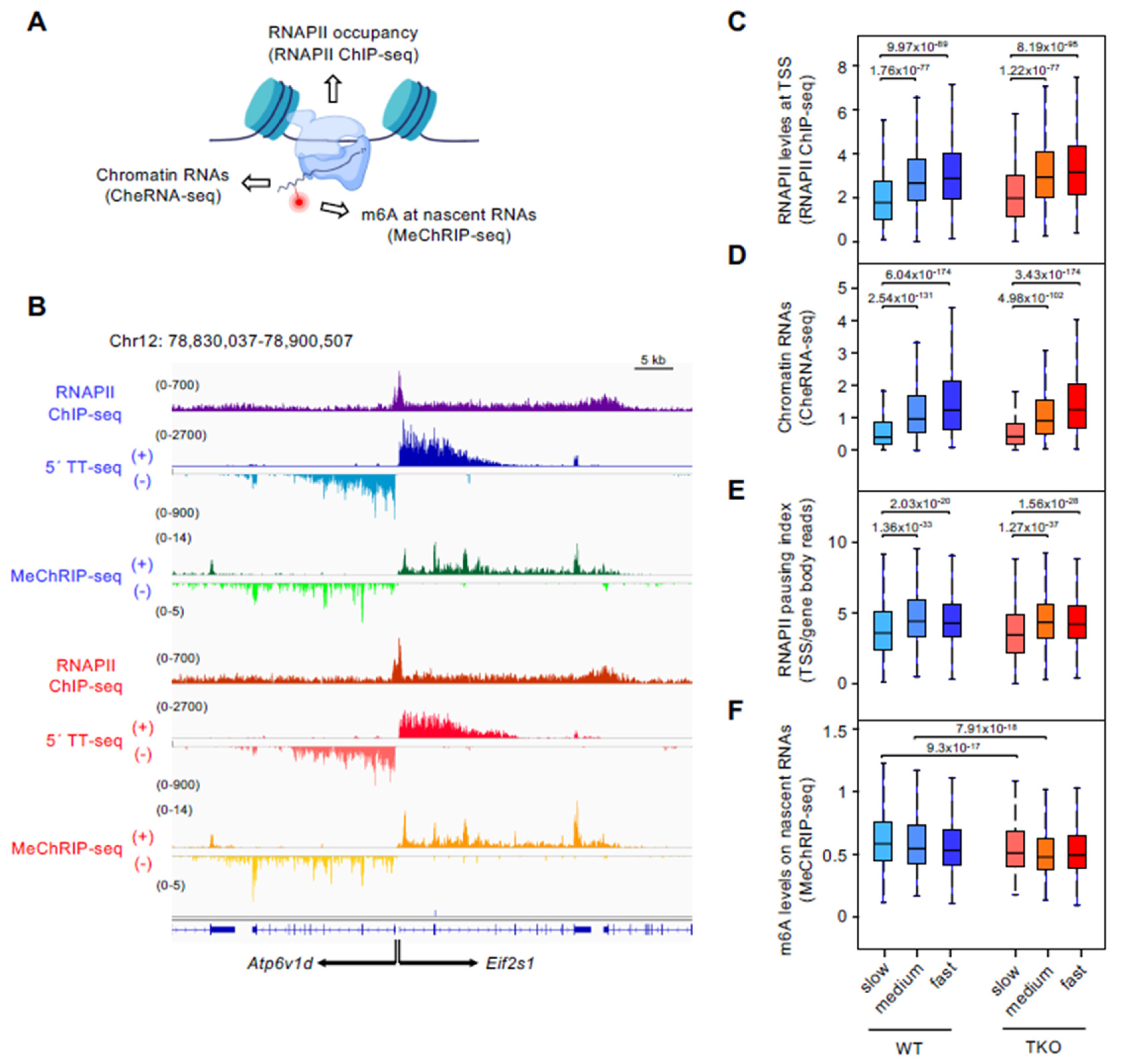
3.2. Slow-Transcribed mRNAs Display Low RNAPII Pausing and High m6A Levels, and Histone H1 Depletion Strongly Decreases Their m6A Deposition
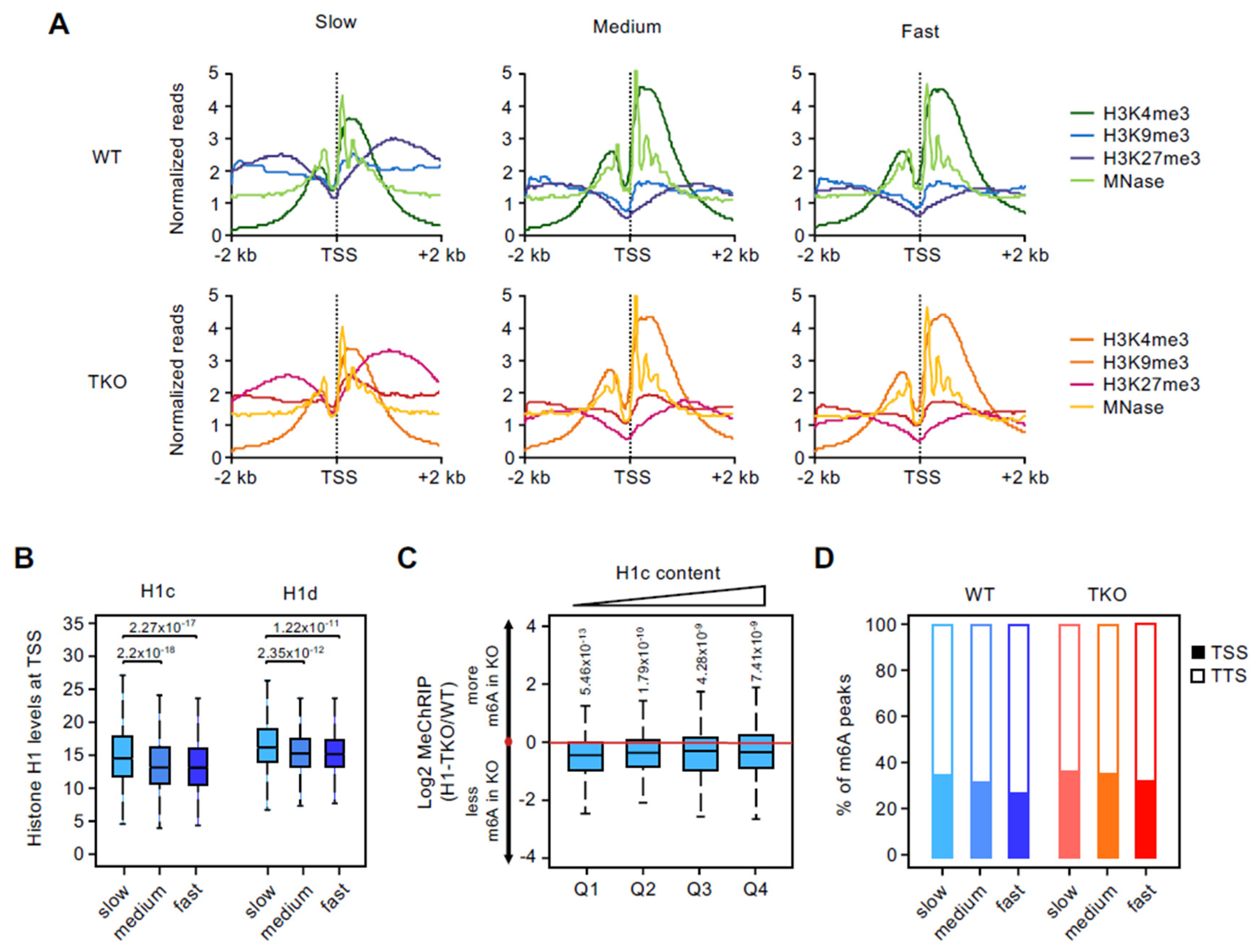
3.3. Promoters of Slow-Rate Genes Are Marked by High Levels of Histone H1 and H3K27me3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic Transcriptomic M6A Decoration: Writers, Erasers, Readers and Functions in RNA Metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by M6A-Dependent Translation Control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 Interactome Reveals That TGFβ Controls M6A MRNA Methylation in Pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 Trimethylation at Lysine 36 Guides M6A RNA Modification Co-Transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef]
- Akhtar, J.; Renaud, Y.; Albrecht, S.; Ghavi-Helm, Y.; Roignant, J.-Y.; Silies, M.; Junion, G. M6A RNA Methylation Regulates Promoter- Proximal Pausing of RNA Polymerase II. Mol. Cell 2021, 81, 3356–3367.e6. [Google Scholar] [CrossRef]
- Xu, W.; He, C.; Kaye, E.G.; Li, J.; Mu, M.; Nelson, G.M.; Dong, L.; Wang, J.; Wu, F.; Shi, Y.G.; et al. Dynamic Control of Chromatin-Associated M6A Methylation Regulates Nascent RNA Synthesis. Mol. Cell 2022, 82, 1156–1168.e7. [Google Scholar] [CrossRef]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.A.F.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the Efficiency of MRNA Translation via Co-Transcriptional N6-Adenosine Methylation. Cell 2017, 169, 326–337.e12. [Google Scholar] [CrossRef]
- Fan, Y.; Nikitina, T.; Zhao, J.; Fleury, T.J.; Bhattacharyya, R.; Bouhassira, E.E.; Stein, A.; Woodcock, C.L.; Skoultchi, A.I. Histone H1 Depletion in Mammals Alters Global Chromatin Structure but Causes Specific Changes in Gene Regulation. Cell 2005, 123, 1199–1212. [Google Scholar] [CrossRef]
- Fernández-Justel, J.M.; Santa-María, C.; Martín-Vírgala, S.; Ramesh, S.; Ferrera-Lagoa, A.; Salinas-Pena, M.; Isoler-Alcaraz, J.; Maslon, M.M.; Jordan, A.; Cáceres, J.F.; et al. Histone H1 Regulates non-coding RNA Turnover on Chromatin in a m6A-dependent Manner. Cell Rep. 2022, 40, 111329. [Google Scholar] [CrossRef]
- Schwalb, B.; Michel, M.; Zacher, B.; Frühauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P. TT-Seq Maps the Human Transient Transcriptome. Science 2016, 352, 1225–1228. [Google Scholar] [CrossRef]
- Maslon, M.M.; Braunschweig, U.; Aitken, S.; Mann, A.R.; Kilanowski, F.; Hunter, C.J.; Blencowe, B.J.; Kornblihtt, A.R.; Adams, I.R.; Cáceres, J.F. A Slow Transcription Rate Causes Embryonic Lethality and Perturbs Kinetic Coupling of Neuronal Genes. EMBO J. 2019, 38, e101244. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Fuchs, G.; Voichek, Y.; Rabani, M.; Benjamin, S.; Gilad, S.; Amit, I.; Oren, M. Simultaneous Measurement of Genome-Wide Transcription Elongation Speeds and Rates of RNA Polymerase II Transition into Active Elongation with 4sUDRB-Seq. Nat. Protoc. 2015, 10, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Voichek, Y.; Benjamin, S.; Gilad, S.; Amit, I.; Oren, M. 4sUDRB-Seq: Measuring Genomewide Transcriptional Elongation Rates and Initiation Frequencies within Cells. Genome Biol. 2014, 15, R69. [Google Scholar] [CrossRef]
- Chen, K.; Xi, Y.; Pan, X.; Li, Z.; Kaestner, K.; Tyler, J.; Dent, S.; He, X.; Li, W. DANPOS: Dynamic Analysis of Nucleosome Position and Occupancy by Sequencing. Genome Res. 2013, 23, 341–351. [Google Scholar] [CrossRef]
- Quintales, L.; Vázquez, E.; Antequera, F. Comparative Analysis of Methods for Genome-Wide Nucleosome Cartography. Brief. Bioinform. 2015, 16, 576–587. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Wei, G.; Almeida, M.; Pintacuda, G.; Coker, H.; Bowness, J.S.; Ule, J.; Brockdorff, N. Acute Depletion of METTL3 Implicates N6-Methyladenosine in Alternative Intron/Exon Inclusion in the Nascent Transcriptome. Genome Res. 2021, 31, 1395–1408. [Google Scholar] [CrossRef] [PubMed]
- Geeven, G.; Zhu, Y.; Kim, B.J.; Bartholdy, B.A.; Yang, S.-M.; Macfarlan, T.S.; Gifford, W.D.; Pfaff, S.L.; Verstegen, M.J.A.M.; Pinto, H.; et al. Local Compartment Changes and Regulatory Landscape Alterations in Histone H1-Depleted Cells. Genome Biol. 2015, 16, 289. [Google Scholar] [CrossRef]
- Cao, K.; Lailler, N.; Zhang, Y.; Kumar, A.; Uppal, K.; Liu, Z.; Lee, E.K.; Wu, H.; Medrzycki, M.; Pan, C.; et al. High-Resolution Mapping of H1 Linker Histone Variants in Embryonic Stem Cells. PLoS Genet. 2013, 9, e1003417. [Google Scholar] [CrossRef] [PubMed]
- Rädle, B.; Rutkowski, A.J.; Ruzsics, Z.; Friedel, C.C.; Koszinowski, U.H.; Dölken, L. Metabolic Labeling of Newly Transcribed RNA for High Resolution Gene Expression Profiling of RNA Synthesis, Processing and Decay in Cell Culture. J. Vis. Exp. 2013, 78, 50195. [Google Scholar] [CrossRef]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Mantsoki, A.; Devailly, G.; Joshi, A. Dynamics of Promoter Bivalency and RNAP II Pausing in Mouse Stem and Differentiated Cells. BMC Dev. Biol. 2018, 18, 2. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego, A.; Fernández-Justel, J.M.; Martín-Vírgala, S.; Maslon, M.M.; Gómez, M. Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs. Genes 2022, 13, 1652. https://doi.org/10.3390/genes13091652
Gallego A, Fernández-Justel JM, Martín-Vírgala S, Maslon MM, Gómez M. Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs. Genes. 2022; 13(9):1652. https://doi.org/10.3390/genes13091652
Chicago/Turabian StyleGallego, Alicia, José Miguel Fernández-Justel, Sara Martín-Vírgala, Magdalena M. Maslon, and María Gómez. 2022. "Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs" Genes 13, no. 9: 1652. https://doi.org/10.3390/genes13091652
APA StyleGallego, A., Fernández-Justel, J. M., Martín-Vírgala, S., Maslon, M. M., & Gómez, M. (2022). Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs. Genes, 13(9), 1652. https://doi.org/10.3390/genes13091652