
Enhanced Resolution of Evolution and Phylogeny of the Moths Inferred from Nineteen Mitochondrial Genomes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. Mitochondrial Genome Sequencing, Assembly, Annotation
2.3. Comparative Analysis of Mitogenome
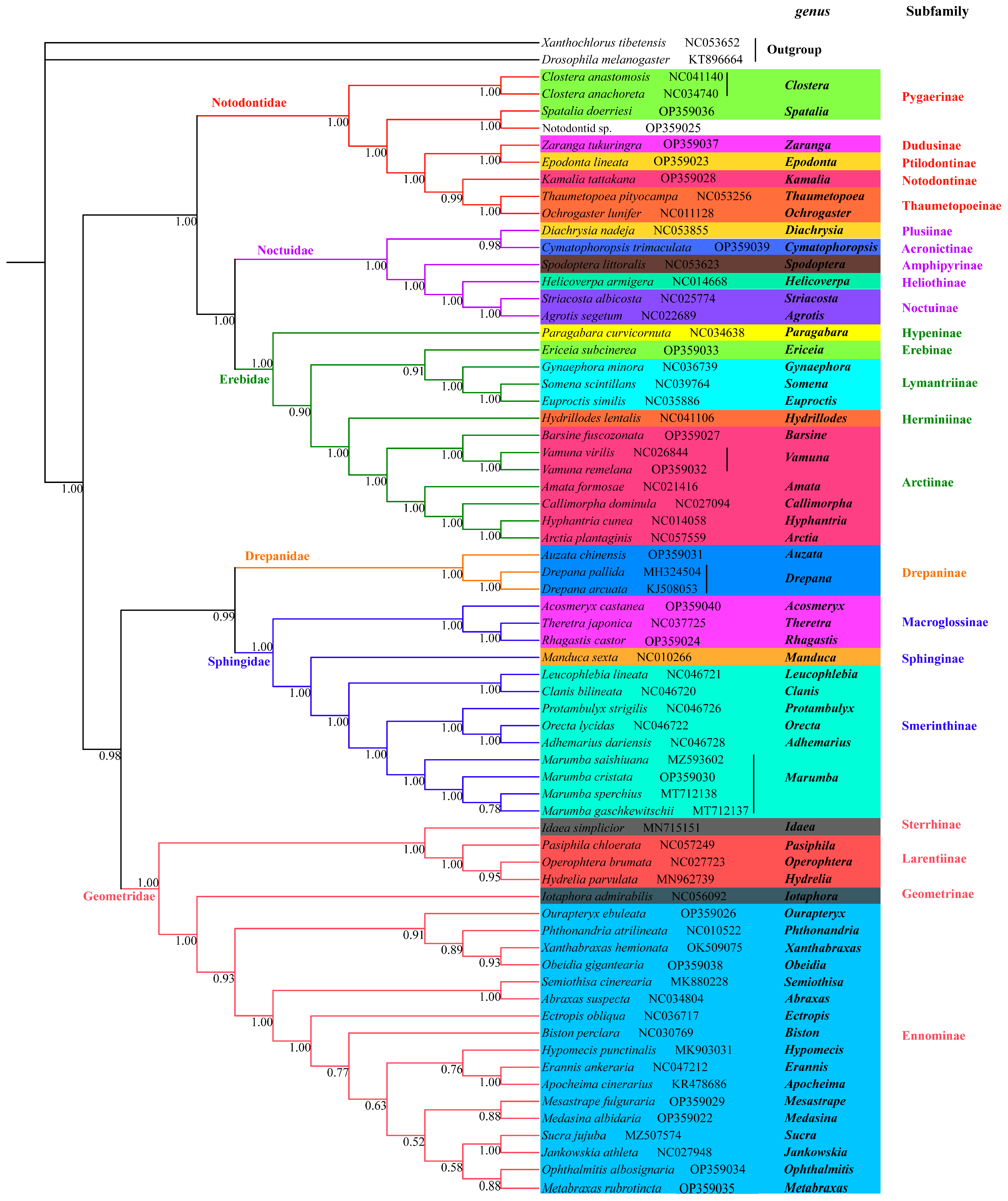
2.4. Phylogenetic Analysis
3. Results and Discussion
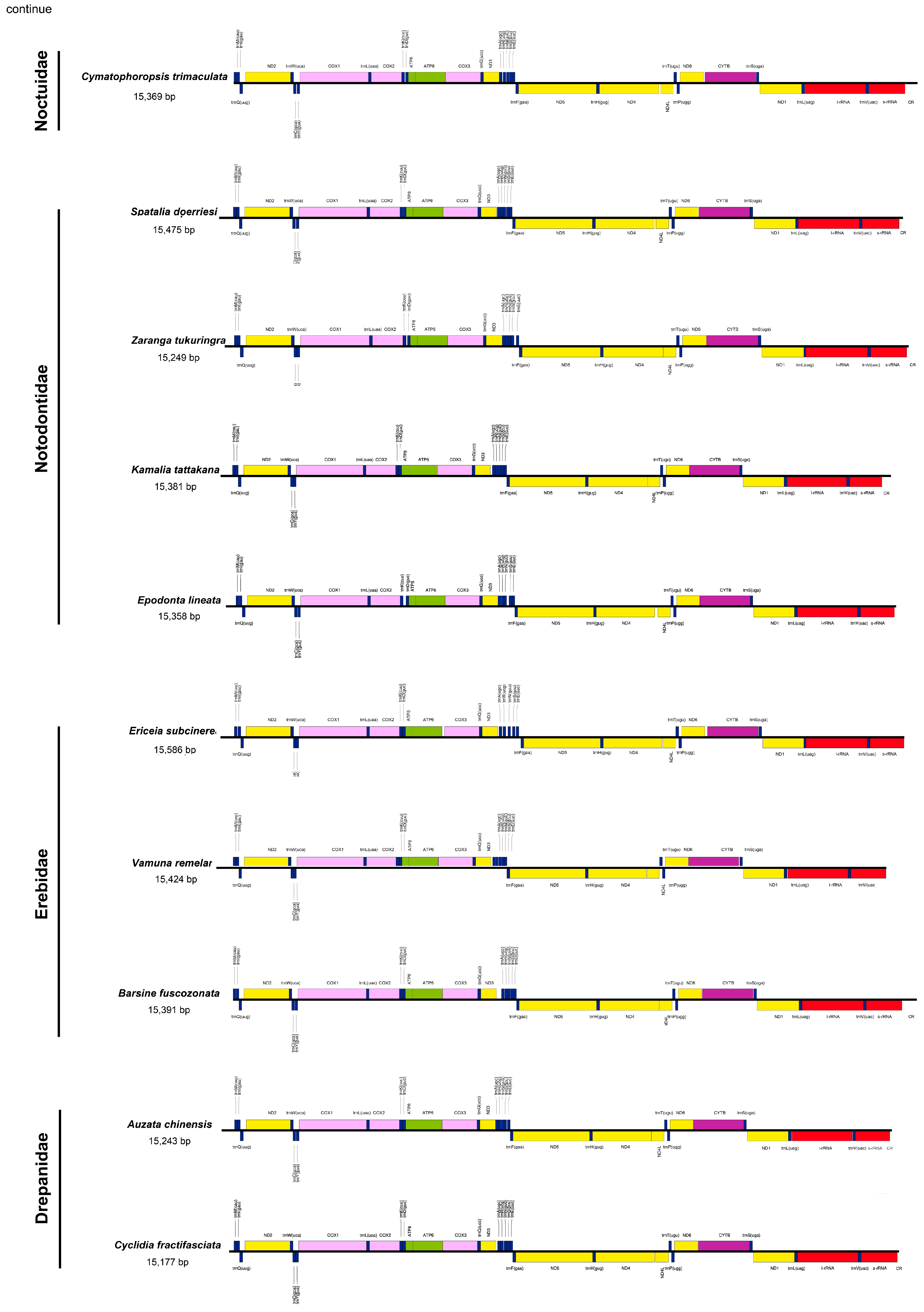
3.1. Mitogenome Structure and Organization
3.2. Overlapping Sequences and Intergenic Spacers
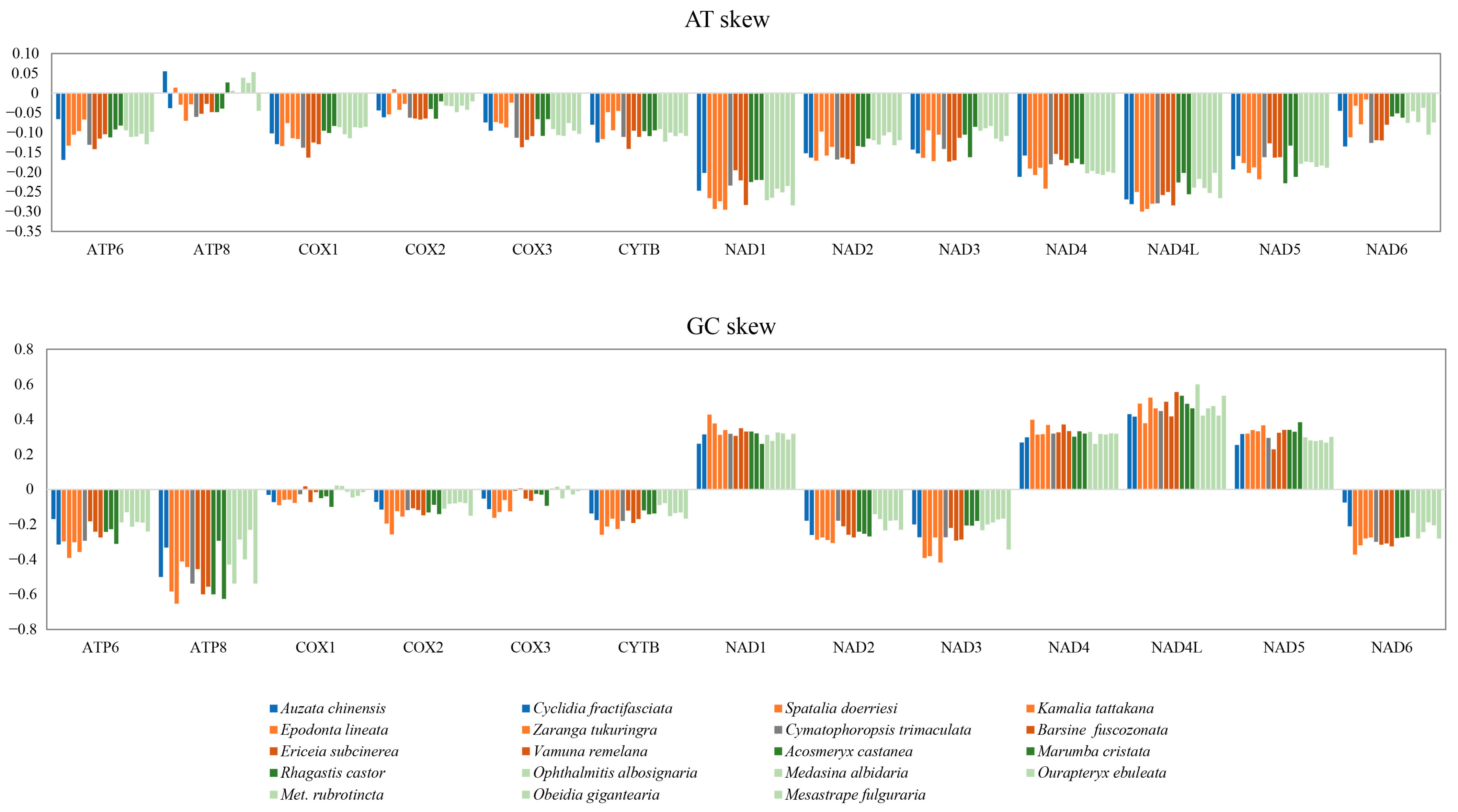
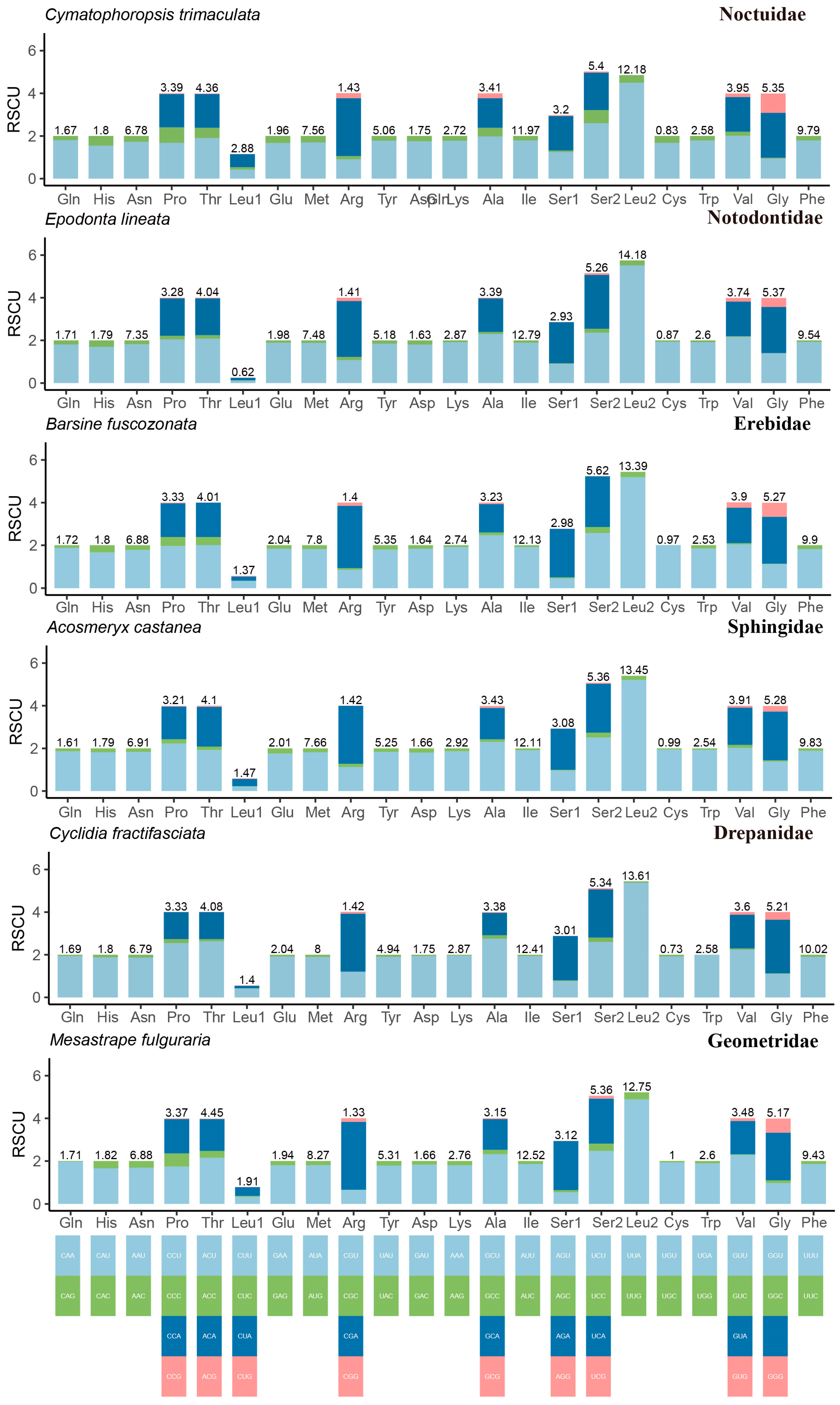
3.3. Codon Usage and Contrasting Rates of Evolution
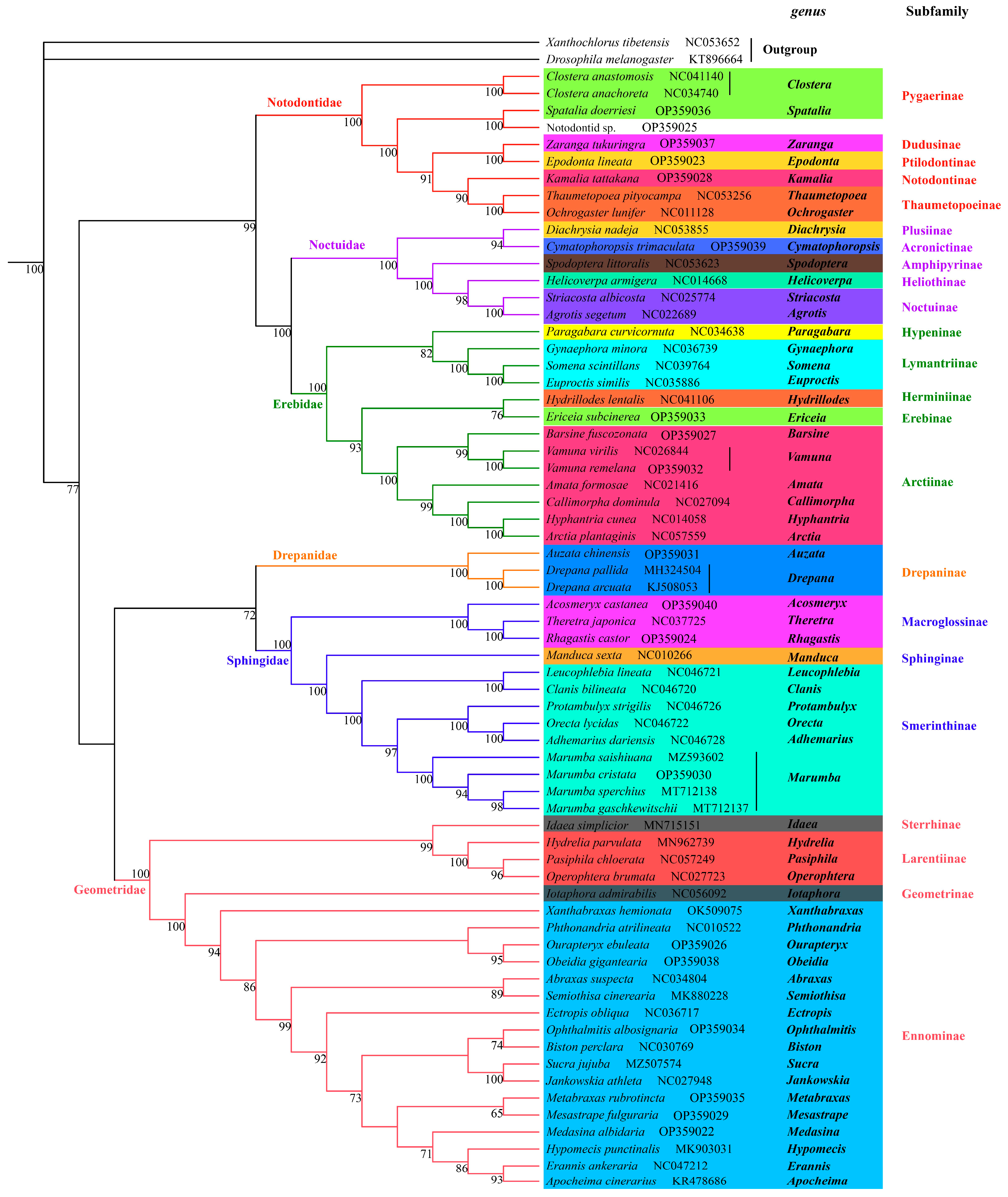
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawahara, A.Y.; Plotkin, D.; Espeland, M.; Meusemann, K.; Toussaint, E.F.; Donath, A.; Gimnich, F.; Frandsen, P.B.; Zwick, A.; Dos Reis, M. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl. Acad. Sci. USA 2019, 116, 22657–22663. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Wheat, C.W.; Peña, C. Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). PLoS ONE 2013, 8, e80875. [Google Scholar] [CrossRef] [PubMed]
- Mitter, C.; Davis, D.R.; Cummings, M.P. Phylogeny and evolution of Lepidoptera. Annu. Rev. Entomol. 2017, 62, 265–283. [Google Scholar] [CrossRef]
- Huseth, A.S.; Koch, R.L.; Reisig, D.; Davis, J.A.; Paula-Moraes, S.V.; Hodgson, E.W. Current distribution and population persistence of five lepidopteran pests in US soybean. J. Integr. Pest Manag. 2021, 12, 11. [Google Scholar] [CrossRef]
- Arditti, J.; Elliott, J.; Kitching, I.J.; Wasserthal, L.T. ‘Good Heavens what insect can suck it’–Charles Darwin, Angraecum sesquipedale and Xanthopan morganii praedicta. Bot. J. Linn. Soc. 2012, 169, 403–432. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, F.; Chen, K. Silkworm: A promising model organism in life science. J. Insect Sci. 2017, 17, 97. [Google Scholar]
- Brady, D.; Saviane, A.; Cappellozza, S.; Sandrelli, F. The Circadian Clock in Lepidoptera. Front. Physiol. 2021, 12, 2049. [Google Scholar] [CrossRef]
- Roe, A.; Weller, S.; Baixeras, J.; Brown, J.; Cummings, M.; Davis, D.; Kawahara, A.; Parr, C.; Regier, J.; Rubinoff, D. Evolutionary framework for Lepidoptera model systems. In Genetics and Molecular Biology of Lepidoptera; CRC Press: Boca Raton, FL, USA, 2009; pp. 1–24. [Google Scholar]
- Ghanavi, H.R.; Twort, V.; Hartman, T.J.; Zahiri, R.; Wahlberg, N. The (non) accuracy of mitochondrial genomes for family level phylogenetics: The case of erebid moths (Lepidoptera; Erebidae). bioRxiv 2021. [Google Scholar] [CrossRef]
- Zahiri, R.; Holloway, J.D.; Kitching, I.J.; Lafontaine, J.D.; Mutanen, M.; Wahlberg, N. Molecular phylogenetics of Erebidae (Lepidoptera, noctuoidea). Syst. Entomol. 2012, 37, 102–124. [Google Scholar] [CrossRef]
- Kergoat, G.J.; Goldstein, P.Z.; Le Ru, B.; Meagher Jr, R.L.; Zilli, A.; Mitchell, A.; Clamens, A.-L.; Gimenez, S.; Barbut, J.; Nègre, N. A novel reference dated phylogeny for the genus Spodoptera Guenée (Lepidoptera: Noctuidae: Noctuinae): New insights into the evolution of a pest-rich genus. Mol. Phylogenetics Evol. 2021, 161, 107161. [Google Scholar] [CrossRef]
- Li, Z.; Wang, W.; Zhang, L. Complete mitochondrial genome of Spodoptera littoralis (Lepidoptera: Noctuidae) from Egypt. Mitochondrial DNA Part B 2021, 6, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Mitter, C.; Regier, J.C. Systematics and evolution of the cutworm moths (Lepidoptera: Noctuidae): Evidence from two protein-coding nuclear genes. Syst. Entomol. 2006, 31, 21–46. [Google Scholar] [CrossRef]
- Rota, J.; Zacharczenko, B.V.; Wahlberg, N.; Zahiri, R.; Schmidt, B.; Wagner, D.L. Phylogenetic relationships of Acronictinae with discussion of the abdominal courtship brush in Noctuidae (Lepidoptera). Syst. Entomol. 2016, 41, 416–429. [Google Scholar] [CrossRef]
- Speidel, W.; Fänger, H.; Naumann, C. The phylogeny of the Noctuidae (Lepidoptera). Syst. Entomol. 1996, 21, 219–252. [Google Scholar] [CrossRef]
- Zahiri, R.; Lafontaine, D.; Schmidt, C.; Holloway, J.D.; Kitching, I.J.; Mutanen, M.; Wahlberg, N. Relationships among the basal lineages of Noctuidae (Lepidoptera, Noctuoidea) based on eight gene regions. Zool. Scr. 2013, 42, 488–507. [Google Scholar] [CrossRef]
- Murillo-Ramos, L.; Brehm, G.; Sihvonen, P.; Hausmann, A.; Holm, S.; Ghanavi, H.R.; Õunap, E.; Truuverk, A.; Staude, H.; Friedrich, E. A comprehensive molecular phylogeny of Geometridae (Lepidoptera) with a focus on enigmatic small subfamilies. PeerJ 2019, 7, e7386. [Google Scholar] [CrossRef]
- Murillo-Ramos, L.; Chazot, N.; Sihvonen, P.; Õunap, E.; Jiang, N.; Han, H.; Clarke, J.T.; Davis, R.B.; Tammaru, T.; Wahlberg, N. Molecular phylogeny, classification, biogeography and diversification patterns of a diverse group of moths (Geometridae: Boarmiini). Mol. Phylogenetics Evol. 2021, 162, 107198. [Google Scholar] [CrossRef]
- Pitkin, L.M. Neotropical ennomine moths: A review of the genera (Lepidoptera: Geometridae). Zool. J. Linn. Soc. 2002, 135, 121–401. [Google Scholar] [CrossRef]
- Sihvonen, P.; Mutanen, M.; Kaila, L.; Brehm, G.; Hausmann, A.; Staude, H.S. Comprehensive molecular sampling yields a robust phylogeny for geometrid moths (Lepidoptera: Geometridae). PLoS ONE 2011, 6, e20356. [Google Scholar] [CrossRef]
- Kristensen, N.P.; Scoble, M.J.; Karsholt, O. Lepidoptera phylogeny and systematics: The state of inventorying moth and butterfly diversity. Zootaxa 2007, 1668, 699–747. [Google Scholar] [CrossRef]
- Kristensen, N.P.; Skalski, A.W. 2. Phylogeny and Palaeontology. Lepid. Moths Butterflies 1998, 1, 7–25. [Google Scholar]
- Minet, J. Ébauche d’une classification modern de l’ordre des Lépidoptères. Alexanor 1986, 14, 291–313. [Google Scholar]
- Minet, J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata). Insect Syst. Evol. 1991, 22, 69–95. [Google Scholar] [CrossRef]
- Mutanen, M.; Wahlberg, N.; Kaila, L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc. R. Soc. B Biol. Sci. 2010, 277, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Regier, J.C.; Cook, C.P.; Mitter, C.; Hussey, A. A phylogenetic study of the ‘bombycoid complex’(Lepidoptera) using five protein-coding nuclear genes, with comments on the problem of macrolepidopteran phylogeny. Syst. Entomol. 2008, 33, 175–189. [Google Scholar] [CrossRef]
- Regier, J.C.; Mitter, C.; Zwick, A.; Bazinet, A.L.; Cummings, M.P.; Kawahara, A.Y.; Sohn, J.-C.; Zwickl, D.J.; Cho, S.; Davis, D.R. A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies). PLoS ONE 2013, 8, e58568. [Google Scholar] [CrossRef]
- Weller, S.; Friedlander, T.; Martin, J.; Pashley, D. Phylogenetic studies of ribosomal RNA variation in higher moths and butterflies (Lepidoptera: Ditrysia). Mol. Phylogenetics Evol. 1992, 1, 312–337. [Google Scholar] [CrossRef]
- Zahiri, R.; Kitching, I.J.; Lafontaine, J.D.; Mutanen, M.; Kaila, L.; Holloway, J.D.; Wahlberg, N. A new molecular phylogeny offers hope for a stable family level classification of the Noctuoidea (Lepidoptera). Zool. Scr. 2011, 40, 158–173. [Google Scholar] [CrossRef]
- Lafontaine, J.D.; Fibiger, M. Revised higher classification of the Noctuoidea (Lepidoptera). Can. Entomol. 2006, 138, 610–635. [Google Scholar] [CrossRef]
- Miller, J.S. Cladistics and Classification of the Notodontidae (Lepidoptera, Noctuoidea) Based on Larval and Adult Morphology. Bulletin of the AMNH; no. 204. 1991. Available online: https://digitallibrary.amnh.org/bitstream/handle/2246/897//v2/dspace/ingest/pdfSource/bul/B204a01.pdf?sequence=1&isAllowed=y (accessed on 30 August 2022).
- Chu, H.; Wang, L. Fauna Sinica. Insecta. Vol. 3. LepidopteraCyclidiidaeDrepanidae; Science Press: Beijing, China, 1991. [Google Scholar]
- Imms, A.D. A General Textbook of Entomology; Dutton: New York, NY, USA, 1934. [Google Scholar]
- Wu, C.; Han, H.; Xue, D. A pilot study on the molecular phylogeny of Drepanoidea (Insecta: Lepidoptera) inferred from the nuclear gene EF-1α and the mitochondrial gene COI. Bull. Entomol. Res. 2010, 100, 207–216. [Google Scholar] [CrossRef]
- Bazinet, A.L.; Cummings, M.P.; Mitter, K.T.; Mitter, C.W. Can RNA-Seq resolve the rapid radiation of advanced moths and butterflies (Hexapoda: Lepidoptera: Apoditrysia)? An exploratory study. PLoS ONE 2013, 8, e82615. [Google Scholar]
- Bian, D.; Ye, W.; Dai, M.; Lu, Z.; Li, M.; Fang, Y.; Qu, J.; Su, W.; Li, F.; Sun, H. Phylogenetic relationships of Limacodidae and insights into the higher phylogeny of Lepidoptera. Int. J. Biol. Macromol. 2020, 159, 356–363. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, H.; Yu, X.; Yang, M. Characterization of two complete mitochondrial genomes of Atkinsoniella (Hemiptera: Cicadellidae: Cicadellinae) and the phylogenetic implications. Insects 2021, 12, 338. [Google Scholar] [CrossRef]
- Zhang, R.; Li, J.; Geng, S.; Yang, J.; Zhang, X.; An, Y.; Li, C.; Cui, H.; Li, X.; Wang, Y. The first mitochondrial genome for Phaudidae (Lepidoptera) with phylogenetic analyses of Zygaenoidea. Int. J. Biol. Macromol. 2020, 149, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Lin, C.-P.; Danforth, B.N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenetics Evol. 2004, 30, 686–702. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, H.; Liu, Y.; Liu, S.; Xia, J.; Zhang, K.; Geng, J. Organization and phylogenetic relationships of the mitochondrial genomes of Speiredonia retorta and other lepidopteran insects. Sci. Rep. 2021, 11, 2957. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, A.R.; Jeong, H.C.; Kim, K.G.; Kim, I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol. Phylogenetics Evol. 2011, 61, 436–445. [Google Scholar] [CrossRef]
- Tian, L.; Sun, X.; Chen, M.; Gai, Y.; Hao, J.; Yang, Q. Complete mitochondrial genome of the Five-dot Sergeant Parathyma sulpitia(Nymphalidae: Limenitidinae) and its phylogenetic implications. Zool. Res. 2012, 33, 133–143. [Google Scholar] [CrossRef]
- Yang, L.; Wei, Z.-J.; Hong, G.-Y.; Jiang, S.-T.; Wen, L.-P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol. Biol. Rep. 2009, 36, 1441–1449. [Google Scholar] [CrossRef]
- Nielsen, E. Phylogeny of Major Lepidopteran Groups. In The Hierarchy of Life; Elsevier: Amsterdam, The Netherlands, 1989; pp. 281–294. [Google Scholar]
- Weller, S.; Pashley, D. In search of butterfly origins. Mol. Phylogenetics Evol. 1995, 4, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, H.; Zhao, T. Insights into the phylogeny of Hemiptera from increased mitogenomic taxon sampling. Mol. Phylogenetics Evol. 2019, 137, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dai, J.; Gao, Q.; Yuan, G.; Liu, J.; Sun, Y.; Sun, Y.; Wang, L.; Qian, C.; Zhu, B. Characterization of the complete mitochondrial genome of Orthaga olivacea Warre (Lepidoptera Pyralidae) and comparison with other Lepidopteran insects. PLoS ONE 2020, 15, e0227831. [Google Scholar] [CrossRef] [PubMed]
- Allio, R.; Scornavacca, C.; Nabholz, B.; Clamens, A.L.; Sperling, F.A.; Condamine, F.L. Whole Genome Shotgun Phylogenomics Resolves the Pattern and Timing of Swallowtail Butterfly Evolution. Syst Biol 2020, 69, 38–60. [Google Scholar] [CrossRef]
- Xue, D. Fauna Sinica, Insecta XV. Lepidoptera; Institute of Zoology, Chinese Academy of Sciences: Beijing, China, 2000. [Google Scholar]
- Zhang, W. A Great Ecological Guide to Insects in China. Chongqing Municipality; Chongqing University Press Co., Ltd.: Chongqing, China, 2011. [Google Scholar]
- Wang, H.; Zhang, X.; Wang, L.; Yue, B.; Feng, S.; Chen, B.; Meng, Y. DNA Barcoding-Based Identification of Geometridae in the Laojunshan National Nature Reserve, Sichuan. Sichuan J. Zool. 2021, 40, 404–414. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 August 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Yang, M.; Song, L.; Zhou, L.; Shi, Y.; Song, N.; Zhang, Y. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: Papilionoidea). Int. J. Biol. Macromol. 2020, 145, 272–281. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, Z.; Yin, J.; Zhang, T.; Zhang, X.; Yuan, D.; Li, T.; Zhong, Y.; Ma, E.; Ren, Z. Complete mitochondrial genome of two Thitarodes species (Lepidoptera, Hepialidae), the host moths of Ophiocordyceps sinensis and phylogenetic implications. Int. J. Biol. Macromol. 2019, 140, 794–807. [Google Scholar] [CrossRef]
- Chai, H.-N.; Du, Y.-Z.; Zhai, B.-P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci. 2012, 8, 561. [Google Scholar] [CrossRef]
- Wei, S.-J.; Shi, M.; Chen, X.-X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.-Y.; He, J.-H. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Kausar, S.; Abbas, M.N.; Wang, T. Complete sequence and characterization of the Ectropis oblique mitochondrial genome and its phylogenetic implications. Int. J. Biol. Macromol. 2018, 107, 1142–1150. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Zhu, X.; Xin, Z.; Zhang, H.; Zhang, D.; Wang, J.; Tang, B.; Zhou, C.; Liu, Q. Comparative mitochondrial genome analysis of Grammodes geometrica and other noctuid insects reveals conserved mitochondrial genome organization and phylogeny. Int. J. Biol. Macromol. 2019, 125, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xin, Z.; Liu, Y.; Wang, Y.; Huang, Y.; Yang, Z.; Chu, X.; Zhang, D.; Zhang, H.; Zhou, C. The complete mitochondrial genome of Clostera anastomosis (Lepidoptera: Notodontidae) and implication for the phylogenetic relationships of Noctuoidea species. Int. J. Biol. Macromol. 2018, 118, 1574–1583. [Google Scholar] [CrossRef]
- Boore, J.L. The Duplication/Random Loss Model for Gene Rearrangement Exemplified by Mitochondrial Genomes of Deuterostome Animals. In Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families; Sankoff, D., Nadeau, J.H., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar]
- Riyaz, M.; Shah, R.A.; Savarimuthu, I.; Kuppusamy, S. Comparative mitochondrial genome analysis of Eudocima salaminia (Cramer, 1777)(Lepidoptera: Noctuoidea), novel gene rearrangement and phylogenetic relationship within the superfamily Noctuoidea. Mol. Biol. Rep. 2021, 48, 4449–4463. [Google Scholar] [CrossRef]
- Cao, Y.-Q.; Ma, C.; Chen, J.-Y.; Yang, D.-R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in Lepidoptera. BMC Genom. 2012, 13, 276. [Google Scholar] [CrossRef]
- Wang, A.R.; Jeong, H.C.; Han, Y.S.; Kim, I. The complete mitochondrial genome of the mountainous duskywing, Erynnis montanus(Lepidoptera: Hesperiidae): A new gene arrangement in Lepidoptera. Mitochondrial DNA 2013, 25, 93–94. [Google Scholar] [CrossRef]
- Doublet, V.; Ubrig, E.; Alioua, A.; Bouchon, D.; Marcadé, I.; Maréchal-Drouard, L. Large gene overlaps and tRNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biol. 2015, 12, 1159–1168. [Google Scholar] [CrossRef]
- Liu, S.Q.; Gu, X.S.; Wang, X. The complete mitochondrial genome of Bombyx lemeepauli (Lepidoptera: Bombycidae) and its phylogenetic relationship. Mitochondrial DNA. Part B Resour. 2017, 2, 518–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Shin, K.S.; Kim, M.S.; Park, H.; Cho, S.; Kim, C.B. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 2006, 373, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, L.; Pape, T.; Gao, Y.; Zhang, D. Evolutionary insights into bot flies (Insecta: Diptera: Oestridae) from comparative analysis of the mitochondrial genomes. Int. J. Biol. Macromol. 2020, 149, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Xu, W.; Zhang, D.; Li, J. Comparative analysis of the mitochondrial genomes of flesh flies and their evolutionary implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef]
- Zahiri, R.; Lafontaine, J.D.; Holloway, J.D.; Kitching, I.J.; Schmidt, B.C.; Kaila, L.; Wahlberg, N. Major lineages of Nolidae (Lepidoptera, Noctuoidea) elucidated by molecular phylogenetics. Cladistics 2013, 29, 337–359. [Google Scholar] [CrossRef]
- Regier, J.C.; Mitter, C.; Mitter, K.; Cummings, M.P.; Bazinet, A.L.; Hallwachs, W.; Janzen, D.H.; Zwick, A. Further progress on the phylogeny of N octuoidea (I nsecta: L epidoptera) using an expanded gene sample. Syst. Entomol. 2017, 42, 82–93. [Google Scholar] [CrossRef]
- Yang, X.; Cameron, S.L.; Lees, D.C.; Xue, D.; Han, H. A mitochondrial genome phylogeny of owlet moths (Lepidoptera: Noctuoidea), and examination of the utility of mitochondrial genomes for lepidopteran phylogenetics. Mol. Phylogenetics Evol. 2015, 85, 230–237. [Google Scholar] [CrossRef]
- Schintlmeister, A. Palaearctic Macrolepidoptera, Volume 1: Notodontidae; Apollo Books: Stenstrup, Denmark, 2008. [Google Scholar]
- Chen, Y. Phylogenetic Reconstruction of Cyclidiinae (Lepidoptera: Drepanidae). Master’s Thesis, National Sun Yat-sen University, Kaohsiung, Taiwan, 2011. [Google Scholar]
- Jiang, N.; Liu, S.; Xue, D.; Han, H. A review of Cyclidiinae from China (Lepidoptera, Drepanidae). ZooKeys 2016, 119. [Google Scholar]
- Schmidt, B.C.; Lafontaine, J.D. Annotated check list of the Noctuoidea (Insecta, Lepidoptera) of North America North of Mexico; PenSoft Publishers LTD: SofiaBu, Bulgaria, 2010; Volume 40. [Google Scholar]
- Heikkilä, M.; Mutanen, M.; Wahlberg, N.; Sihvonen, P.; Kaila, L. Elusive ditrysian phylogeny: An account of combining systematized morphology with molecular data (Lepidoptera). BMC Evol. Biol. 2015, 15, 260. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Kitching, I.; Xu, Z.-B.; Huang, Y.-X. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene 2021, 789, 145667. [Google Scholar] [CrossRef] [PubMed]
- Holloway, J. The moths ofBomeo: Family Geometridae: Subfamilies Sterrhinae, Larentiinae. Malay. Nat. J. 1997, 51, 1–242. [Google Scholar]
- Ounap, E.; Viidalepp, J.; Truuverk, A. Phylogeny of the subfamily L arentiinae (L epidoptera: G eometridae): Integrating molecular data and traditional classifications. Syst. Entomol. 2016, 41, 824–843. [Google Scholar] [CrossRef]
- Yamamoto, S.; Sota, T. Phylogeny of the Geometridae and the evolution of winter moths inferred from a simultaneous analysis of mitochondrial and nuclear genes. Mol. Phylogenetics Evol. 2007, 44, 711–723. [Google Scholar] [CrossRef]
- Sihvonen, P.; Kaila, L. Phylogeny and tribal classification of Sterrhinae with emphasis on delimiting Scopulini (Lepidoptera: Geometridae). Syst. Entomol. 2004, 29, 324–358. [Google Scholar] [CrossRef]
- Ounap, E.; JavoiŠ, J.; Viidalepp, J.; Tammaru, T. Phylogenetic relationships of selected European Ennominae (Lepidoptera: Geometridae). Eur. J. Entomol. 2011, 108, 267–273. [Google Scholar] [CrossRef]
- Wahlberg, N.; Snäll, N.; Viidalepp, J.; Ruohomäki, K.; Tammaru, T. The evolution of female flightlessness among Ennominae of the Holarctic forest zone (Lepidoptera, Geometridae). Mol. Phylogenetics Evol. 2010, 55, 929–938. [Google Scholar] [CrossRef]
- Young, C.J. Molecular relationships of the Australian Ennominae (Lepidoptera: Geometridae) and implications for the phylogeny of the Geometridae from molecular and morphological data. Zootaxa 2006, 1264, 1–147. [Google Scholar] [CrossRef]
- Holloway, J.D. The moths of Borneo: Family Geometridae, subfamily Ennominae. [=The Moths of Borneo, Part 11]. Malay. Nat. J. 1994, 47, 1–19. [Google Scholar]
- Viidalepp, J.; Tammaru, T.; Snäll, N.; Ruohomäki, K.; Wahlberg, N. Cleorodes Warren, 1894 does not belong in the tribe Boarmiini (Lepidoptera: Geometridae). Eur. J. Entomol. 2007, 104, 303. [Google Scholar] [CrossRef]
- Stekolnikov, A.; Kuznetzov, V. Functional morphology of the male genitalia and arrangement of new species of the geometrid moths: Subfamily Ennominae (Lepidoptera, Geometridae). Entomol. Obozr. 1982, 59, 344–374. [Google Scholar]
- Sihvonen, P.; Staude, H.S.; Mutanen, M. Systematic position of the enigmatic African cycad moths: An integrative approach to a nearly century old problem (Lepidoptera: Geometridae, Diptychini). Syst. Entomol. 2015, 40, 606–627. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Mitogenome Length (bp) | A% | T (U)% | C% | G% | AT (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|
| Drepanidae | Auzata chinensis | 15,243 | 41.1 | 39.2 | 11.60 | 8.20 | 80.30 | 19.80 |
| Cyc. fractifasciata | 15,177 | 39.8 | 40.8 | 11.90 | 7.50 | 80.60 | 19.40 | |
| Erebidae | B. fuscozonata | 15,391 | 40.2 | 41.7 | 10.70 | 7.40 | 81.90 | 18.10 |
| Ericeia subcinerea | 15,586 | 39.9 | 40.3 | 12.40 | 7.50 | 80.20 | 19.90 | |
| Vamuna remelana | 15,424 | 40.2 | 40.2 | 12.10 | 7.50 | 80.40 | 19.60 | |
| Geometridae | Medasina albidaria | 15,504 | 41.4 | 39.8 | 10.90 | 7.90 | 81.20 | 18.80 |
| Mesastrape fulguraria | 15,561 | 41.7 | 39.4 | 11.40 | 7.40 | 81.10 | 18.80 | |
| Metabraxas rubrotincta | 15,577 | 41.9 | 39.5 | 11.20 | 7.50 | 81.40 | 18.70 | |
| Obeidia gigantearia | 15,429 | 41.2 | 39.9 | 11.20 | 7.70 | 81.10 | 18.90 | |
| Op. albosignaria | 15,749 | 42.1 | 39.8 | 10.70 | 7.40 | 81.90 | 18.10 | |
| Ourapteryx ebuleata | 15,667 | 41 | 39.5 | 11.70 | 7.80 | 80.50 | 19.50 | |
| Noctuidae | Cymatophoropsis trimaculata | 15,369 | 40.1 | 40.3 | 11.80 | 7.70 | 80.40 | 19.50 |
| Notodontidae | Epodonta lineata | 15,358 | 40.7 | 39.3 | 12.20 | 7.70 | 80.00 | 19.90 |
| K. tattakana | 15,381 | 40.7 | 37.4 | 13.80 | 8.10 | 78.10 | 21.90 | |
| Spatalia doerriesi | 15,475 | 39.2 | 39.7 | 13.50 | 7.70 | 78.90 | 21.20 | |
| Zaranga tukuringra | 15,249 | 41.1 | 37.5 | 13.60 | 7.90 | 78.60 | 21.50 | |
| Sphingidae | Acosmeryx castanea | 15,201 | 41 | 39.6 | 11.80 | 7.60 | 80.60 | 19.40 |
| Marumba cristata | 15,744 | 40.5 | 41.1 | 11.10 | 7.30 | 81.60 | 18.40 | |
| Rhagastis castor | 15,265 | 41.1 | 39.2 | 12.30 | 7.50 | 80.30 | 19.80 |
| Species | AT Skew | GC Skew | ||||||
|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 123 | 1st | 2nd | 3rd | 123 | |
| Au. chinensis | −0.0052 | −0.3718 | −0.0616 | −0.1373 | 0.2430 | −0.0965 | −0.1821 | 0.0303 |
| Cyc. fractifasciata | 0.0023 | −0.3761 | −0.0902 | −0.1461 | 0.1934 | −0.0988 | −0.2555 | 0.0007 |
| B. fuscozonata | −0.0046 | −0.3766 | −0.0856 | −0.1466 | 0.2484 | −0.1102 | −0.2340 | 0.0376 |
| Er. subcinerea | −0.0054 | −0.3707 | −0.0822 | −0.1448 | 0.2408 | −0.1057 | −0.2493 | 0.0165 |
| V. remelana | −0.0095 | −0.3698 | −0.0949 | −0.1496 | 0.2451 | −0.1096 | −0.2853 | 0.0205 |
| Med. albidaria | −0.0126 | −0.3665 | −0.0660 | −0.1388 | 0.2639 | −0.0947 | −0.1465 | 0.0533 |
| Met. rubrotincta | 0.0002 | −0.3622 | −0.0647 | −0.1331 | 0.2907 | −0.1043 | −0.2281 | 0.0447 |
| Ma. cristata | −0.0065 | −0.3652 | −0.0423 | −0.1268 | 0.2560 | −0.0925 | −0.2909 | 0.0351 |
| Op. albosignaria | −0.0053 | −0.3598 | −0.0636 | −0.1334 | 0.2933 | −0.0963 | −0.1317 | 0.0634 |
| Ob. gigantearia | −0.0224 | −0.3607 | −0.0606 | −0.1371 | 0.2772 | −0.0984 | −0.2333 | 0.0369 |
| Ou. ebuleata | −0.0078 | −0.3642 | −0.0642 | −0.1366 | 0.2732 | −0.0921 | −0.2059 | 0.0359 |
| Cym. trimaculata | −0.0155 | −0.3735 | −0.0901 | −0.1512 | 0.2457 | −0.0980 | −0.2776 | 0.0203 |
| Ep. lineata | −0.0125 | −0.3743 | −0.0825 | −0.1482 | 0.2350 | −0.1044 | −0.2757 | 0.0117 |
| K. tattakana | 0.0122 | −0.3690 | −0.0577 | −0.1305 | 0.1908 | −0.1142 | −0.2283 | −0.0228 |
| S. doerriesi | −0.0014 | −0.3740 | −0.0994 | −0.1522 | 0.1919 | −0.0971 | −0.2088 | −0.0049 |
| Mes. fulguraria | −0.0078 | −0.3613 | −0.0792 | −0.1400 | 0.2594 | −0.1028 | −0.2616 | 0.0204 |
| Z. tukuringra | 0.0062 | −0.3739 | −0.0776 | −0.1410 | 0.1906 | −0.1063 | −0.2044 | −0.0056 |
| Ac. castanea | −0.0215 | −0.3668 | −0.0582 | −0.1381 | 0.2508 | −0.1036 | −0.1589 | 0.0383 |
| Ma. cristata | −0.0065 | −0.3652 | −0.0423 | −0.1268 | 0.2560 | −0.0925 | −0.2909 | 0.0351 |
| R. castor | 0.0174 | −0.3668 | −0.0550 | −0.1255 | 0.2039 | −0.1048 | −0.2125 | 0.0037 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Zhang, R.; Yue, B.; Wu, Y.; Yang, N.; Zhou, C. Enhanced Resolution of Evolution and Phylogeny of the Moths Inferred from Nineteen Mitochondrial Genomes. Genes 2022, 13, 1634. https://doi.org/10.3390/genes13091634
Zheng X, Zhang R, Yue B, Wu Y, Yang N, Zhou C. Enhanced Resolution of Evolution and Phylogeny of the Moths Inferred from Nineteen Mitochondrial Genomes. Genes. 2022; 13(9):1634. https://doi.org/10.3390/genes13091634
Chicago/Turabian StyleZheng, Xiaofeng, Rusong Zhang, Bisong Yue, Yongjie Wu, Nan Yang, and Chuang Zhou. 2022. "Enhanced Resolution of Evolution and Phylogeny of the Moths Inferred from Nineteen Mitochondrial Genomes" Genes 13, no. 9: 1634. https://doi.org/10.3390/genes13091634
APA StyleZheng, X., Zhang, R., Yue, B., Wu, Y., Yang, N., & Zhou, C. (2022). Enhanced Resolution of Evolution and Phylogeny of the Moths Inferred from Nineteen Mitochondrial Genomes. Genes, 13(9), 1634. https://doi.org/10.3390/genes13091634