
GWAS and Transcriptome Analysis Reveal Key Genes Affecting Root Growth under Low Nitrogen Supply in Maize
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Phenotypic Evaluation and Data Analysis
2.3. Genotypic Data and Genome-Wide Association Analysis
2.4. RNA-Seq Analysis of Maize Root under Low N
2.5. Quantitative Real-Time PCR
2.6. Candidate Gene Resequencing and Gene-Based Association Mapping
3. Results
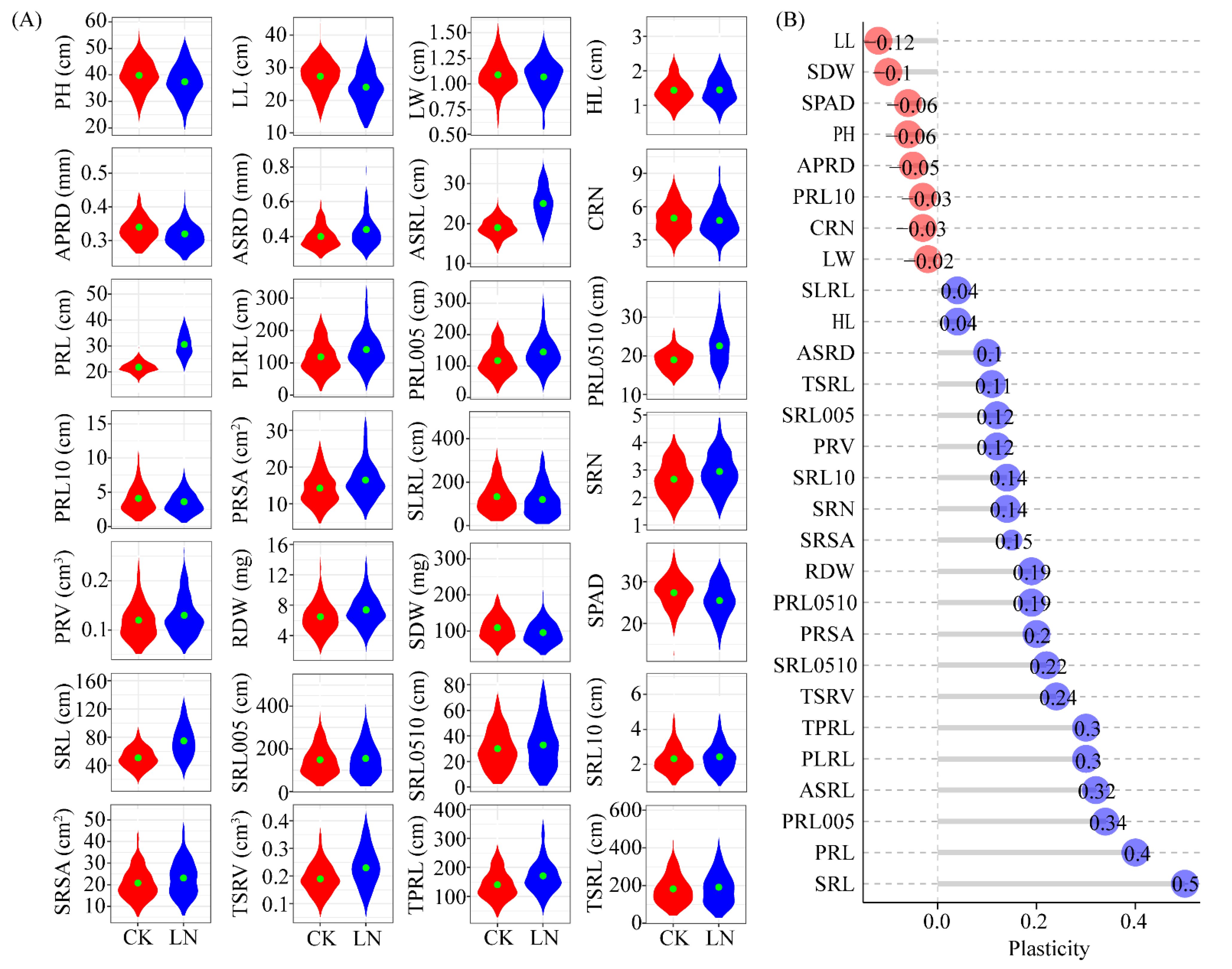
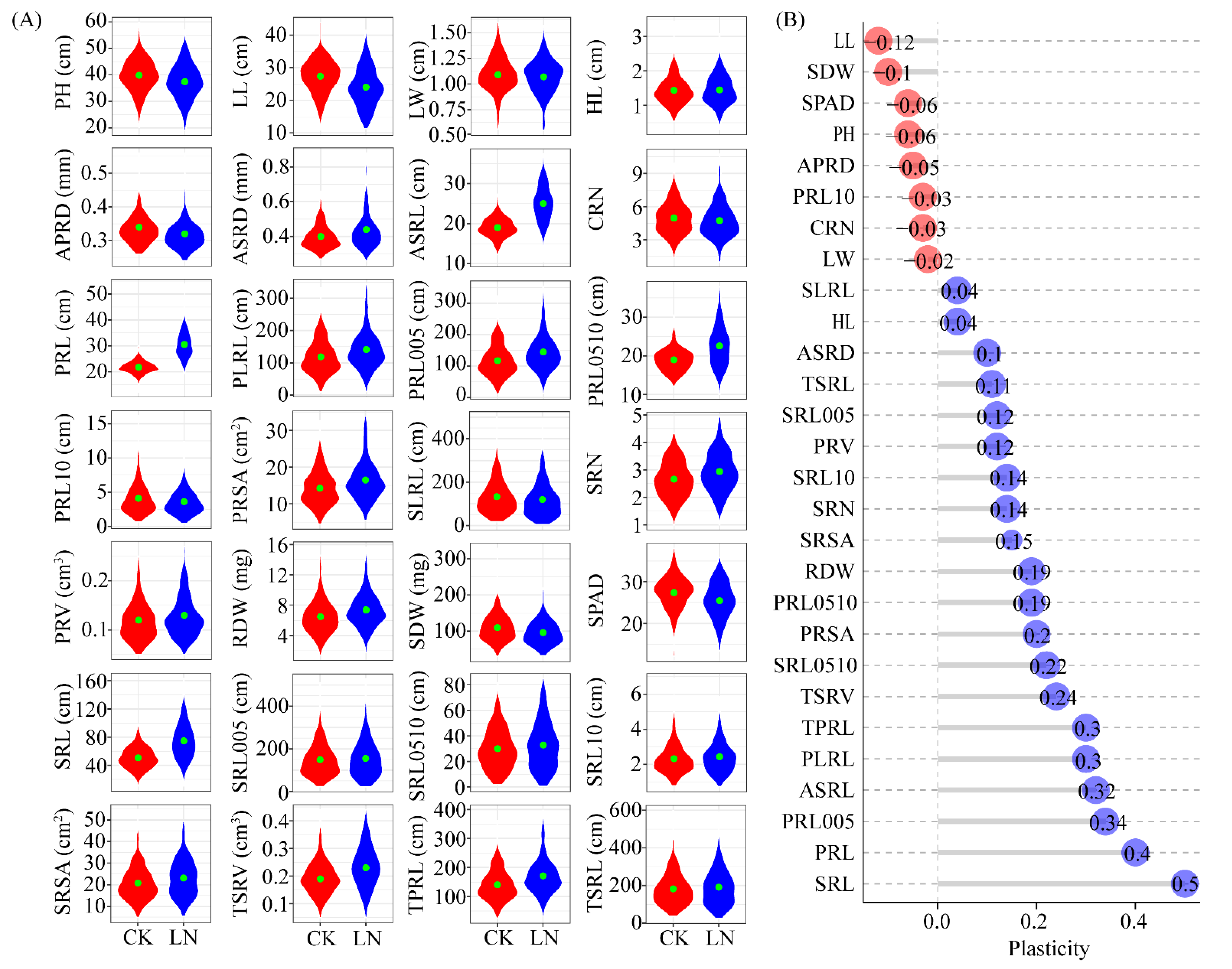
3.1. Root Traits Differences under Normal and Nitrogen-Deficient Conditions
3.2. Association Analysis of the Root Traits under Low-N Conditions
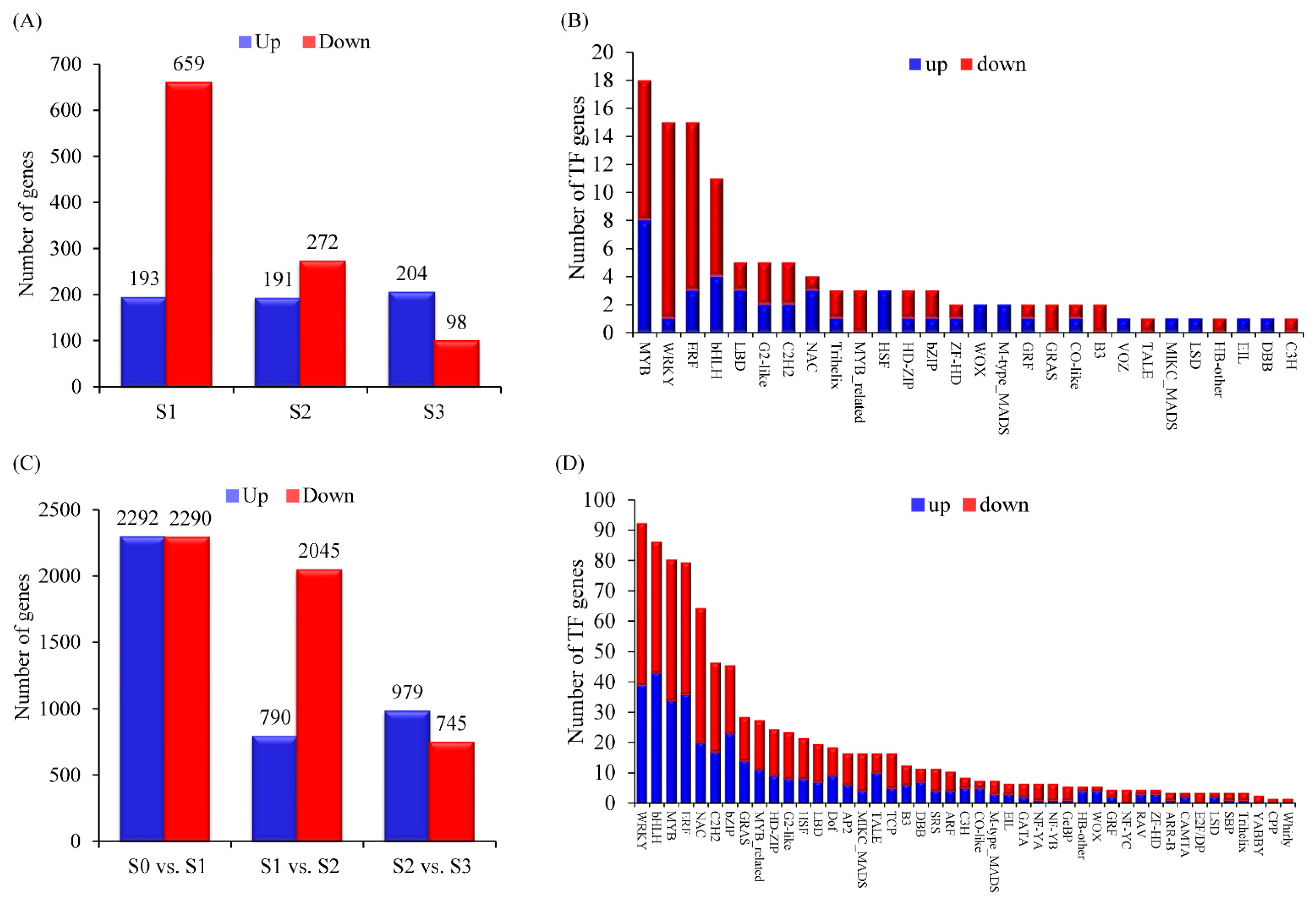
3.3. Whole-Genome Transcriptome Analysis of the B73 Root in Response to a Low-N Supply
3.4. Integrating GWAS and RNA-Seq Data to Prioritize Causal Genes
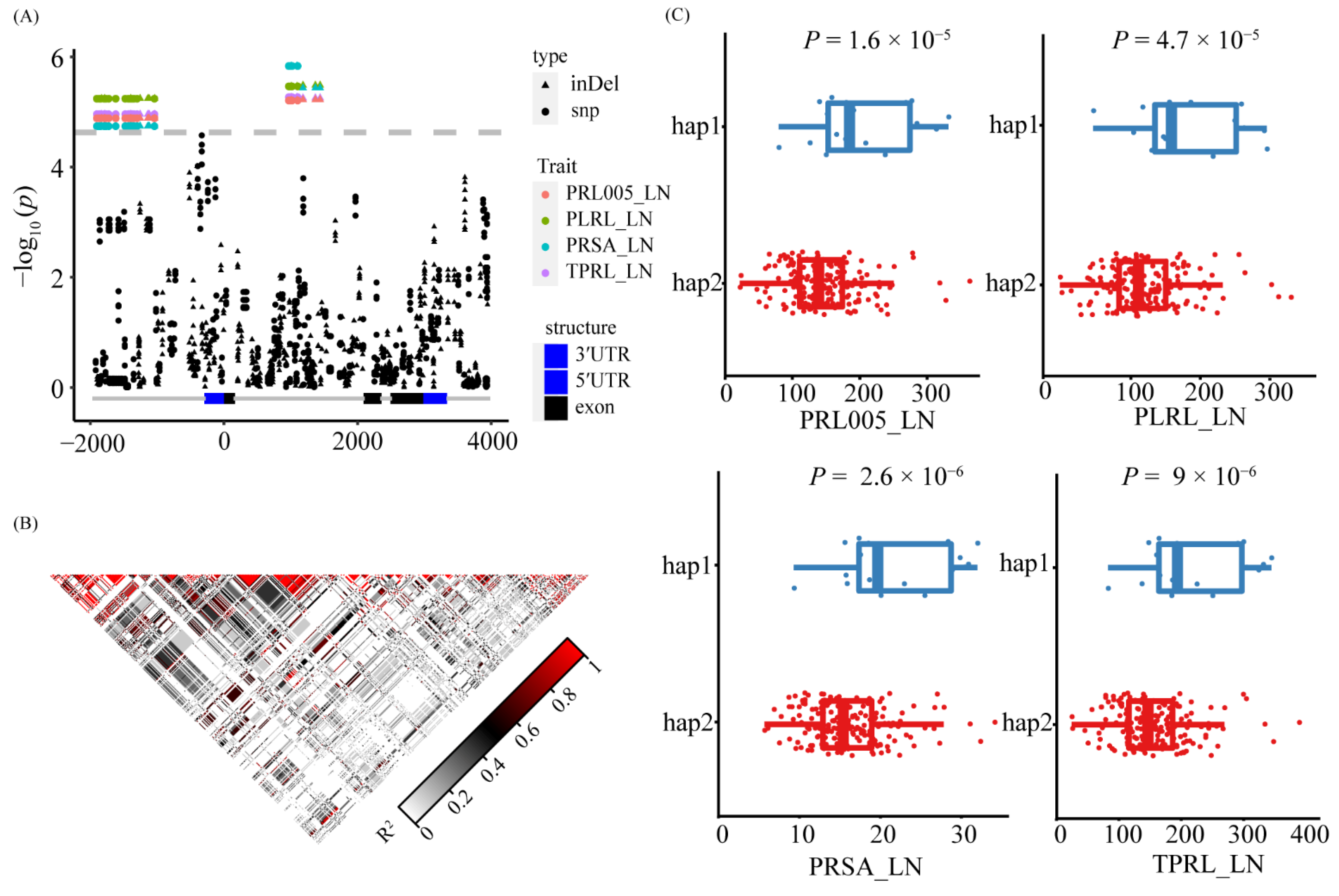
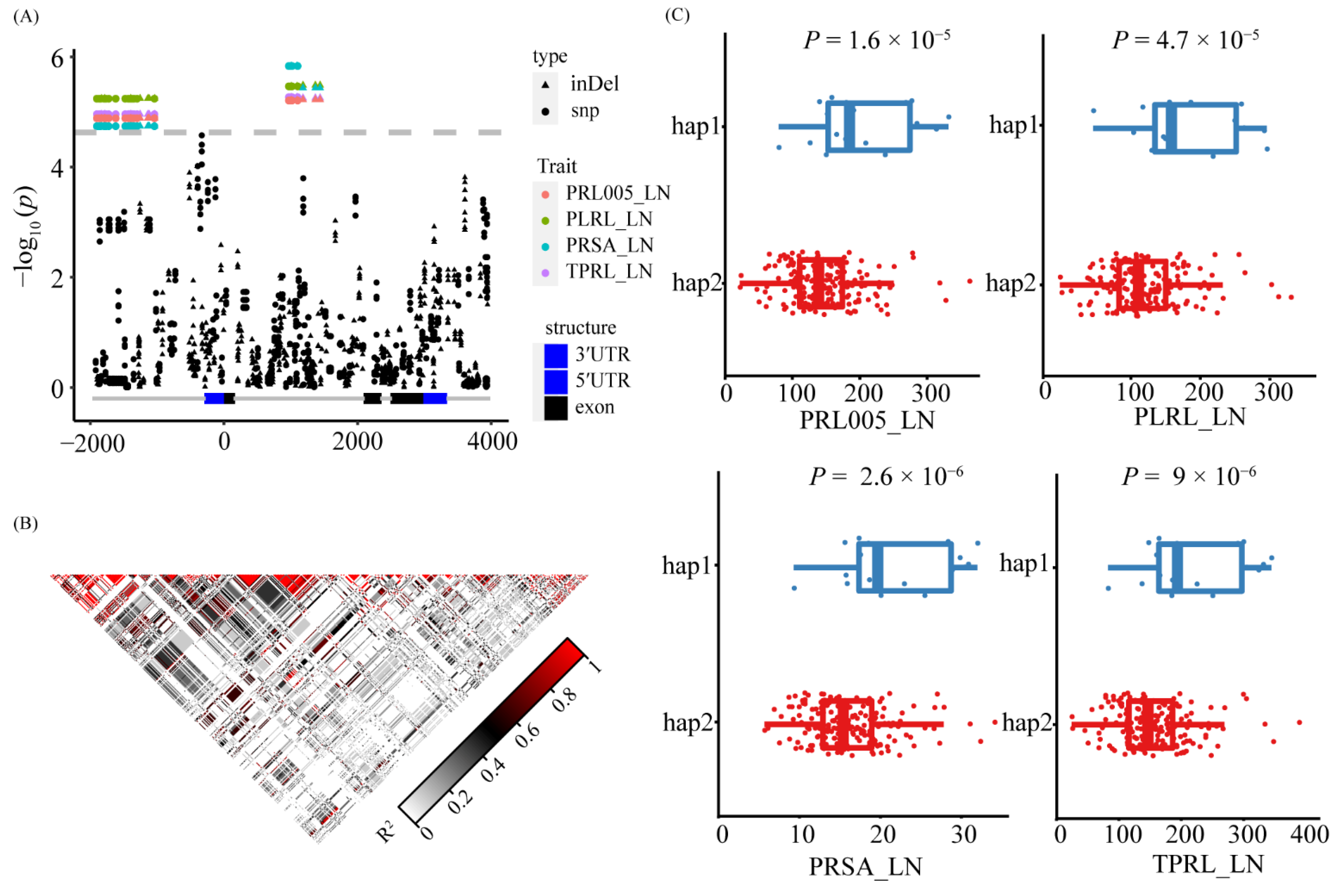
3.5. Natural Variation of ZmNAC36 Affect Root Traits in Response to Nitrogen Deficiency
4. Discussion
4.1. Effects of Low-N Conditions on the Root Architecture
4.2. Candidate Genes Identified in the Detected QTL Regions for Root Traits under Low-N Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wani, S.H.; Vijayan, R.; Choudhary, M.; Kumar, A.; Zaid, A.; Singh, V.; Kumar, P.; Yasin, J.K. Nitrogen use efficiency (NUE): Elucidated mechanisms, mapped genes and gene networks in maize (Zea mays L.). Physiol. Mol. Biol. Plants 2021, 27, 2875–2891. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, B.; Chu, C. Nitrogen assimilation in plants: Current status and future prospects. J. Genet. Genom. 2022, 49, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.N.; Townsend, A.R.; Erisman, J.W.; Bekunda, M.; Cai, Z.; Freney, J.R.; Martinelli, L.A.; Seitzinger, S.P.; Sutton, M.A. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 2008, 320, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.H.; Liu, X.J.; Zhang, Y.; Shen, J.L.; Han, W.X.; Zhang, W.F.; Christie, P.; Goulding, K.W.; Vitousek, P.M.; Zhang, F.S. Significant acidification in major Chinese croplands. Science 2010, 327, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Han, W.; Tang, A.; Shen, J.; Cui, Z.; Vitousek, P.; Erisman, J.W.; Goulding, K.; Christie, P.; et al. Enhanced nitrogen deposition over China. Nature 2013, 494, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P. Steep, cheap and deep: An ideotype to optimize water and N acquisition by maize root systems. Ann. Bot. 2013, 112, 347–357. [Google Scholar] [CrossRef]
- Li, P.; Chen, F.; Cai, H.; Liu, J.; Pan, Q.; Liu, Z.; Gu, R.; Mi, G.; Zhang, F.; Yuan, L. A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J. Exp. Bot. 2015, 66, 3175–3188. [Google Scholar] [CrossRef]
- Rao, I.M.; Miles, J.W.; Beebe, S.E.; Horst, W.J. Root adaptations to soils with low fertility and aluminium toxicity. Ann. Bot. 2016, 118, 593–605. [Google Scholar] [CrossRef]
- Mu, X.; Chen, F.; Wu, Q.; Chen, Q.; Wang, J.; Yuan, L.; Mi, G. Genetic improvement of root growth increases maize yield via enhanced post-silking nitrogen uptake. Eur. J. Agron. 2015, 63, 55–61. [Google Scholar] [CrossRef]
- Hochholdinger, F.; Yu, P.; Marcon, C. Genetic control of root system development in maize. Trends Plant Sci. 2018, 23, 79–88. [Google Scholar] [CrossRef]
- Zhan, A.; Lynch, J.P. Reduced frequency of lateral root branching improves N capture from low-N soils in maize. J. Exp. Bot. 2015, 66, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, A.C.; McClymont, S.A.; Holmes, B.M.; Lyons, E.; Raizada, M.N. Novel temporal, fine-scale and growth variation phenotypes in roots of adult-stage maize (Zea mays L.) in response to low nitrogen stress. Plant Cell Environ. 2011, 34, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhuang, Z.; Cai, H.; Cheng, S.; Soomro, A.A.; Liu, Z.; Gu, R.; Mi, G.; Yuan, L.; Chen, F. Use of genotype-environment interactions to elucidate the pattern of maize root plasticity to nitrogen deficiency. J. Integr. Plant Biol. 2016, 58, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Giehl, R.F.H.; von Wirén, N. The root foraging response under low nitrogen depends on DWARF1-mediated brassinosteroid biosynthesis. Plant Physiol. 2020, 183, 998–1010. [Google Scholar] [CrossRef]
- Sun, C.H.; Yu, J.Q.; Hu, D.G. Nitrate: A crucial signal during lateral roots development. Front. Plant Sci. 2017, 8, 485. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Mounier, E.; Pervent, M.; Ljung, K.; Gojon, A.; Nacry, P. Auxin-mediated nitrate signalling by NRT1.1 participates in the adaptive response of Arabidopsis root architecture to the spatial heterogeneity of nitrate availability. Plant Cell Environ. 2014, 37, 162–174. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, H.; Hamera, S.; Chen, X.; Fang, R. miR444a has multiple functions in the rice nitrate-signaling pathway. Plant J. 2014, 78, 44–55. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Z.; Li, X.; Wang, Z.; Chen, F.; Mi, G.; Forde, B.; Takahashi, H.; Yuan, L. Involvement of a truncated MADS-box transcription factor ZmTMM1 in root nitrate foraging. J. Exp. Bot. 2020, 71, 4547–4561. [Google Scholar] [CrossRef]
- Forde, B.G. Local and long-range signaling pathways regulating plant responses to nitrate. Annu. Rev. Plant Biol. 2002, 53, 203–224. [Google Scholar] [CrossRef]
- Maghiaoui, A.; Bouguyon, E.; Cuesta, C.; Perrine-Walker, F.; Alcon, C.; Krouk, G.; Benková, E.; Nacry, P.; Gojon, A.; Bach, L. The Arabidopsis NRT1.1 transceptor coordinately controls auxin biosynthesis and transport to regulate root branching in response to nitrate. J. Exp. Bot. 2020, 71, 4480–4494. [Google Scholar] [CrossRef] [PubMed]
- Lay-Pruitt, K.S.; Takahashi, H. Integrating N signals and root growth: The role of nitrate transceptor NRT1.1 in auxin-mediated lateral root development. J. Exp. Bot. 2020, 71, 4365–4368. [Google Scholar] [CrossRef] [PubMed]
- Naulin, P.A.; Armijo, G.I.; Vega, A.S.; Tamayo, K.P.; Gras, D.E.; de la Cruz, J.; Gutiérrez, R.A. Nitrate induction of primary root growth requires cytokinin signaling in Arabidopsis thaliana. Plant Cell Physiol. 2020, 61, 342–352. [Google Scholar] [CrossRef]
- Tian, Q.Y.; Sun, P.; Zhang, W.H. Ethylene is involved in nitrate-dependent root growth and branching in Arabidopsis thaliana. New Phytol. 2009, 184, 918–931. [Google Scholar] [CrossRef]
- Signora, L.; De Smet, I.; Foyer, C.H.; Zhang, H. ABA plays a central role in mediating the regulatory effects of nitrate on root branching in Arabidopsis. Plant J. 2001, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Giehl, R.F.H.; von Wirén, N. Local auxin biosynthesis acts downstream of brassinosteroids to trigger root foraging for nitrogen. Nat. Commun. 2021, 12, 5437. [Google Scholar] [CrossRef]
- Jia, Z.; Giehl, R.F.H.; Meyer, R.C.; Altmann, T.; von Wirén, N. Natural variation of BSK3 tunes brassinosteroid signaling to regulate root foraging under low nitrogen. Nat. Commun. 2019, 10, 2378. [Google Scholar] [CrossRef]
- Namai, S.; Toriyama, K.; Fukuta, Y. Genetic variations in dry matter production and physiological nitrogen use efficiency in rice (Oryza sativa L.) varieties. Breed. Sci. 2009, 59, 269–276. [Google Scholar] [CrossRef]
- Hu, B.; Wang, W.; Ou, S.; Tang, J.; Li, H.; Che, R.; Zhang, Z.; Chai, X.; Wang, H.; Wang, Y.; et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef]
- Li, P.; Pan, T.; Wang, H.; Wei, J.; Chen, M.; Hu, X.; Zhao, Y.; Yang, X.; Yin, S.; Xu, Y.; et al. Natural variation of ZmHKT1 affects root morphology in maize at the seedling stage. Planta 2019, 249, 879–889. [Google Scholar] [CrossRef]
- Xu, S.; Tang, X.; Zhang, X.; Wang, H.; Ji, W.; Xu, C.; Yang, Z.; Li, P. Genome-wide association study identifies novel candidate loci or genes affecting stalk strength in maize. Crop J. 2022. [Google Scholar] [CrossRef]
- Wang, H.; Wei, J.; Li, P.; Wang, Y.; Ge, Z.; Qian, J.; Fan, Y.; Ni, J.; Xu, Y.; Yang, Z.; et al. Integrating GWAS and gene expression analysis identifies candidate genes for root morphology traits in maize at the seedling stage. Genes 2019, 10, 773. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, X.; Wang, H.; Pan, T.; Wang, Y.; Xu, Y.; Xu, C.; Yang, Z. Genetic control of root plasticity in response to salt stress in maize. Theor. Appl. Genet. 2021, 134, 1475–1492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mi, G.; Chen, F.; Zhang, J.; Zhang, F. Response of root morphology to nitrate supply and its contribution to nitrogen accumulation in maize. J. Plant Nutr. 2004, 27, 2189–2202. [Google Scholar] [CrossRef]
- Li, M.X.; Yeung, J.M.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2012, 131, 747–756. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P. Variant annotation and functional prediction: SnpEff. Methods Mol. Biol. 2022, 2493, 289–314. [Google Scholar] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2013, 25, 402–408. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.N.; Lee, T.Y.; Hung, Y.C.; Li, G.Z.; Tseng, K.C.; Liu, Y.H.; Kuo, P.L.; Zheng, H.Q.; Chang, W.C. PlantPAN3.0: A new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Res. 2019, 47, D1155–D1163. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Chen, F.; Zhang, F.; Ren, T.; Zhuang, Z.; Mi, G. Mapping QTLs for root traits under different nitrate levels at the seedling stage in maize (Zea mays L.). Plant Soil 2008, 305, 253–265. [Google Scholar] [CrossRef]
- Chen, X.; Cui, Z.; Fan, M.; Vitousek, P.; Zhao, M.; Ma, W.; Wang, Z.; Zhang, W.; Yan, X.; Yang, J.; et al. Producing more grain with lower environmental costs. Nature 2014, 514, 486–489. [Google Scholar] [CrossRef]
- Sun, X.; Ren, W.; Wang, P.; Chen, F.; Yuan, L.; Pan, Q.; Mi, G. Evaluation of maize root growth and genome-wide association studies of root traits in response to low nitrogen supply at seedling emergence. Crop J. 2021, 9, 794–804. [Google Scholar] [CrossRef]
- Schneider, H.M.; Lynch, J.P. Should root plasticity be a crop breeding target? Front. Plant Sci. 2020, 11, 546. [Google Scholar] [CrossRef]
- Kant, S. Understanding nitrate uptake, signaling and remobilisation for improving plant nitrogen use efficiency. Semin. Cell Dev. Biol. 2018, 74, 89–96. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. Regulation of Arabidopsis root development by nitrate availability. J. Exp. Bot. 2000, 51, 51–59. [Google Scholar] [CrossRef]
- McMurtrie, R.E.; Iversen, C.M.; Dewar, R.C.; Medlyn, B.E.; Näsholm, T.; Pepper, D.A.; Norby, R.J. Plant root distributions and nitrogen uptake predicted by a hypothesis of optimal root foraging. Ecol. Evol. 2012, 2, 1235–1250. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, C.; Jiang, Y.; Tian, Y.; Yu, J.; An, H.; Tang, W.; Sun, J.; Tang, J.; Chen, G.; et al. Association mapping and genetic dissection of nitrogen use efficiency-related traits in rice (Oryza sativa L.). Funct. Integr. Genom. 2016, 16, 323. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.M. NIT proteins regulate rice root plasticity in response to nitrate and ammonium. Plant Physiol. 2020, 183, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.G.; Caixeta, D.G.; Rezende, W.M.; Schuster, A.; Azevedo, C.F.; e Silva, F.F.; Delima, R.O. Genotypic variation and relationships among traits for root morphology in a panel of tropical maize inbred lines under contrasting nitrogen levels. Euphytica 2019, 215, 51. [Google Scholar] [CrossRef]
- Wu, B.; Ren, W.; Zhao, L.; Li, Q.; Sun, J.; Chen, F.; Pan, Q. Genome-wide association study of root system architecture in maize. Genes 2022, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, R.J.; Michno, J.M.; Jeffers, J.; Hoekenga, O.; Dilkes, B.; Baxter, I.; Myers, C.L. Integrating coexpression networks with GWAS to prioritize causal genes in maize. Plant Cell 2018, 30, 2922–2942. [Google Scholar] [CrossRef]
- Sekhon, R.S.; Hirsch, C.N.; Childs, K.L.; Breitzman, M.W.; Kell, P.; Duvick, S.; Spalding, E.P.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Phenotypic and transcriptional analysis of divergently selected maize populations reveals the role of developmental timing in seed size determination. Plant Physiol. 2014, 165, 658–669. [Google Scholar] [CrossRef]
- Chang, L.; Ramireddy, E.; Schmülling, T. Cytokinin as a positional cue regulating lateral root spacing in Arabidopsis. J. Exp. Bot. 2015, 66, 4759–4768. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Onckelen, H.V.; Schmülling, T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef]
- Gao, S.; Fang, J.; Xu, F.; Wang, W.; Sun, X.; Chu, J.; Cai, B.; Feng, Y.; Chu, C. CYTOKININ OXIDASE/DEHYDROGENASE4 integrates cytokinin and auxin signaling to control rice crown root formation. Plant Physiol. 2014, 165, 1035–1046. [Google Scholar] [CrossRef]
- Huangfu, L.; Chen, R.; Lu, Y.; Zhang, E.; Miao, J.; Zuo, Z.; Zhao, Y.; Zhu, M.; Zhang, Z.; Li, P.; et al. OsCOMT, encoding a caffeic acid O-methyltransferase in melatonin biosynthesis, increases rice grain yield through dual regulation of leaf senescence and vascular development. Plant Biotechnol. J. 2022, 20, 1122–1139. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, J.; Song, J.; Jameson, P.E. Cytokinin dehydrogenase: A genetic target for yield improvement in wheat. Plant Biotechnol. J. 2020, 18, 614–630. [Google Scholar] [CrossRef] [PubMed]
- Ramireddy, E.; Hosseini, S.A.; Eggert, K.; Gillandt, S.; Gnad, H.; von Wirén, N.; Schmulling, T. Root engineering in barley: Increasing cytokinin degradation produces a larger root system, mineral enrichment in the shoot and improved drought tolerance. Plant Physiol. 2018, 177, 1078–1095. [Google Scholar] [CrossRef] [PubMed]
- Ramireddy, E.; Nelissen, H.; Leuendorf, J.E.; Lijsebettens, M.V.; Inze, D.; Schmulling, T. Root engineering in maize by increasing cytokinin degradation causes enhanced root growth and leaf mineral enrichment. Plant Mol. Biol. 2021, 106, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Waidmann, S.; Rosquete, M.R.; Schöller, M.; Lindner, H.; LaRue, T.; Sarkel, E.; Petřík, I.; Dünser, K.; Martopawiro, S.; Sasidharan, R.; et al. Cytokinin functions as an asymmetric and anti-gravitropic signal in lateral roots. Nat. Commun. 2019, 10, 3540. [Google Scholar] [CrossRef]
- Wang, H.; Sun, H.; Xia, H.; Wu, T.; Li, P.; Xu, C.; Yang, Z. Natural variation and domestication selection of ZmCKX5 with root morphological traits at the seedling stage in maize. Plants 2020, 10, 1. [Google Scholar] [CrossRef]
- Mao, C.; He, J.; Liu, L.; Deng, Q.; Yao, X.; Liu, C.; Qiao, Y.; Li, P.; Ming, F. OsNAC2 integrates auxin and cytokinin pathways to modulate rice root development. Plant Biotechnol. J. 2020, 18, 429–442. [Google Scholar] [CrossRef]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W.; Tian, Y.; Zhang, Y.; Xu, S.; An, H.; Chen, G.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef]
- Xu, P.; Ma, W.; Hu, J.; Cai, W. The nitrate-inducible NAC transcription factor NAC056 controls nitrate assimilation and promotes lateral root growth in Arabidopsis thaliana. PLoS Genet. 2022, 18, e1010090. [Google Scholar] [CrossRef]
- Vidal, E.A.; Moyano, T.C.; Riveras, E.; Contreras-López, O.; Gutiérrez, R.A. Systems approaches map regulatory networks downstream of the auxin receptor AFB3 in the nitrate response of Arabidopsis thaliana roots. Proc. Natl. Acad. Sci. USA 2013, 110, 12840–12845. [Google Scholar] [CrossRef]
- He, X.; Qu, B.; Li, W.; Zhao, X.; Teng, W.; Ma, W.; Ren, Y.; Li, B.; Li, Z.; Tong, Y. The nitrate-inducible NAC transcription factor TaNAC2-5A controls nitrate response and increases wheat yield. Plant Physiol. 2015, 169, 1991–2005. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Mi, Y.; Mao, H.; Liu, S.; Chen, L.; Qin, F. Genetic variation in ZmTIP1 contributes to root hair elongation and drought tolerance in maize. Plant Biotechnol. J. 2020, 18, 1271–1283. [Google Scholar] [PubMed]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.S.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6, 8326. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Li, L.; Li, J.; Zhuang, M.; Li, B.; Li, Q.; Huang, J.; Du, Y.; Wang, J.; et al. TaMOR is essential for root initiation and improvement of root system architecture in wheat. Plant Biotechnol. J. 2022, 20, 862–875. [Google Scholar] [CrossRef]
- Mao, H.; Li, S.; Chen, B.; Jian, C.; Mei, F.; Zhang, Y.; Li, F.; Chen, N.; Li, T.; Du, L.; et al. Variation in cis-regulation of a NAC transcription factor contributes to drought tolerance in wheat. Mol. Plant 2022, 15, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Sun, X.; Gao, S.; Qin, F.; Dai, M. Deletion of an endoplasmic reticulum stress response element in a ZmPP2C-A gene facilitates drought tolerance of maize seedlings. Mol. Plant 2017, 10, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 170, 114–126.e15. [Google Scholar] [CrossRef]
- Ye, J.; Wang, X.; Hu, T.; Zhang, F.; Wang, B.; Li, C.; Yang, T.; Li, H.; Lu, Y.; Giovannoni, J.J.; et al. An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. Plant Cell 2017, 29, 2249–2268. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhu, T.; Yang, J.; Wang, H.; Ji, W.; Xu, Y.; Yang, Z.; Xu, C.; Li, P. GWAS and Transcriptome Analysis Reveal Key Genes Affecting Root Growth under Low Nitrogen Supply in Maize. Genes 2022, 13, 1632. https://doi.org/10.3390/genes13091632
Wang Y, Zhu T, Yang J, Wang H, Ji W, Xu Y, Yang Z, Xu C, Li P. GWAS and Transcriptome Analysis Reveal Key Genes Affecting Root Growth under Low Nitrogen Supply in Maize. Genes. 2022; 13(9):1632. https://doi.org/10.3390/genes13091632
Chicago/Turabian StyleWang, Yunyun, Tianze Zhu, Jiyuan Yang, Houmiao Wang, Weidong Ji, Yang Xu, Zefeng Yang, Chenwu Xu, and Pengcheng Li. 2022. "GWAS and Transcriptome Analysis Reveal Key Genes Affecting Root Growth under Low Nitrogen Supply in Maize" Genes 13, no. 9: 1632. https://doi.org/10.3390/genes13091632
APA StyleWang, Y., Zhu, T., Yang, J., Wang, H., Ji, W., Xu, Y., Yang, Z., Xu, C., & Li, P. (2022). GWAS and Transcriptome Analysis Reveal Key Genes Affecting Root Growth under Low Nitrogen Supply in Maize. Genes, 13(9), 1632. https://doi.org/10.3390/genes13091632