_Haendeler.png)
Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and DNA Isolation
2.3. Biochemical Analysis
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. Clinical and Biochemical Analysis
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lam, C.S.; Roger, V.L.; Rodeheffer, R.J.; Borlaug, B.A.; Enders, F.T.; Redfield, M.M. Pulmonary hypertension in heart failure with preserved ejection fraction: A community-based study. J. Am. Coll. Cardiol. 2009, 53, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.; Lafont, A.; Limongelli, G.; Mahrholdt, H. Authors/Task Force members. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Bloemink, M.; Deacon, J.; Langer, S.; Vera, C.; Combs, A.; Leinwand, L.; Geeves, M.A. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin. J. Biol. Chem. 2014, 289, 5158–5167. [Google Scholar] [CrossRef]
- Kensler, R.W.; Shaffer, J.F.; Harris, S.P. Binding of the N-terminal fragment C0–C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 2011, 174, 44–51. [Google Scholar] [CrossRef]
- Burkett, E.L.; Hershberger, R.E. Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981. [Google Scholar] [CrossRef]
- Palmer, B.M. Thick filament proteins and performance in human heart failure. Heart Fail. Rev. 2005, 10, 187–197. [Google Scholar] [CrossRef]
- Walsh, R.; Rutland, C.; Thomas, R.; Loughna, S. Cardiomyopathy: A systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010, 115, 49–60. [Google Scholar]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E. A molecular basis for familial Hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Fananapazir, L.; Dalakas, M.C.; Cyran, F.; Cohn, G.; Epstein, N.D. Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 1993, 90, 3993–3997. [Google Scholar] [CrossRef]
- Hesaraki, M.; Bora, U.; Pahlavan, S.; Salehi, N.; Mousavi, S.A.; Barekat, M.; Rasouli, S.J.; Baharvand, H.; Ozhan, G.; Totonchi, M. A Novel Missense Variant in Actin Binding Domain of MYH7 Is Associated With Left Ventricular Noncompaction. Front. Cardiovasc. Med. 2022, 9, 839862. [Google Scholar] [CrossRef] [PubMed]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef]
- Dufour, C.; Dausse, E.; Fetler, L.; Dubourg, O.; Bouhour, J.B.; Vosberg, H.P.; Guicheney, P.; Komajda, M.; Schwartz, K. Identification of a mutation near a functional site of the beta cardiac myosin heavy chain gene in a family with hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 1994, 26, 1241–1247. [Google Scholar] [CrossRef]
- Daehmlow, S.; Erdmann, J.; Knueppel, T.; Gille, C.; Froemmel, C.; Hummel, M.; Hetzer, R.; Regitz-Zagrosek, V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 298, 116–120. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.W.; Lee, J.; Kim, Y. Genetic variations leading to familial dilated cardiomyopathy. Mol. Cells 2016, 39, 722. [Google Scholar]
- Dellefave, L.; McNally, E.M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 198. [Google Scholar] [CrossRef]
- Ujfalusi, Z.; Vera, C.D.; Mijailovich, S.M.; Svicevic, M.; Yu, E.C.; Kawana, M.; Ruppel, K.M.; Spudich, J.A.; Geeves, M.A.; Leinwand, L.A. Dilated cardiomyopathy myosin mutants have reduced force-generating capacity. J. Biol. Chem. 2018, 293, 9017–9029. [Google Scholar] [CrossRef]
- Lakdawala, N.K.; Thune, J.J.; Colan, S.D.; Cirino, A.L.; Farrohi, F.; Rivero, J.; McDonough, B.; Sparks, E.; Orav, E.; Seidman, J. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2012, 5, 503–510. [Google Scholar] [CrossRef]
- Debold, E.P.; Schmitt, J.P.; Patlak, J.; Beck, S.; Moore, J.; Seidman, J.G.; Seidman, C.; Warshaw, D.M. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H284–H291. [Google Scholar] [CrossRef]
- Palmer, B.M.; Schmitt, J.P.; Seidman, C.E.; Seidman, J.; Wang, Y.; Bell, S.P.; LeWinter, M.M.; Maughan, D.W. Elevated rates of force development and MgATP binding in F764L and S532P myosin mutations causing dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 57, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Debold, E.P.; Ahmad, F.; Armstrong, A.; Frederico, A.; Conner, D.A.; Mende, U.; Lohse, M.J.; Warshaw, D.; Seidman, C.E. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. USA 2006, 103, 14525–14530. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
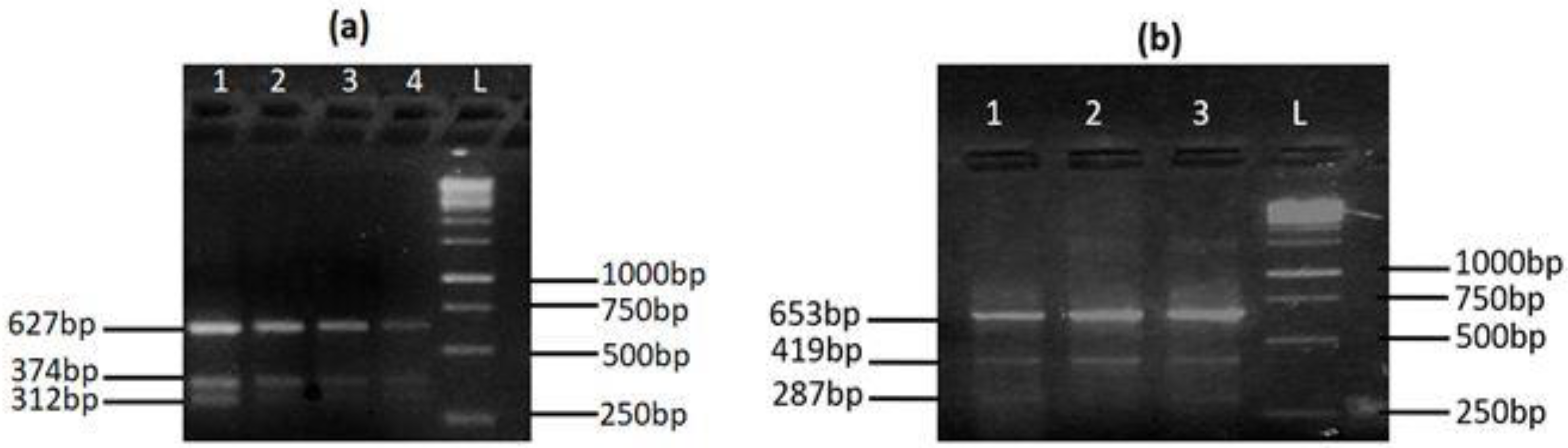
| Primers | Primers Sequences (5′–3′) | Tm (°C) | Product Size |
|---|---|---|---|
| MYH7rs121913642 | |||
| Forward outer | CCTAGCATCTCAGGCATCTGGGTCGTGGAGTG | 66.8 | Control = 627 bp T allele = 374 bp C allele = 312 bp |
| Reverse outer | CCTTGGCAGAAACCCTGCTCCTCTGTACCG | 66.5 | |
| Forward inner | CCTGCTTCCTCAGCACATGGGCATCAGGT | 67.1 | |
| Reverse inner | CCTTGGGGAACATGCAGTCCTCTTCCAGGATTGG | 66.9 | |
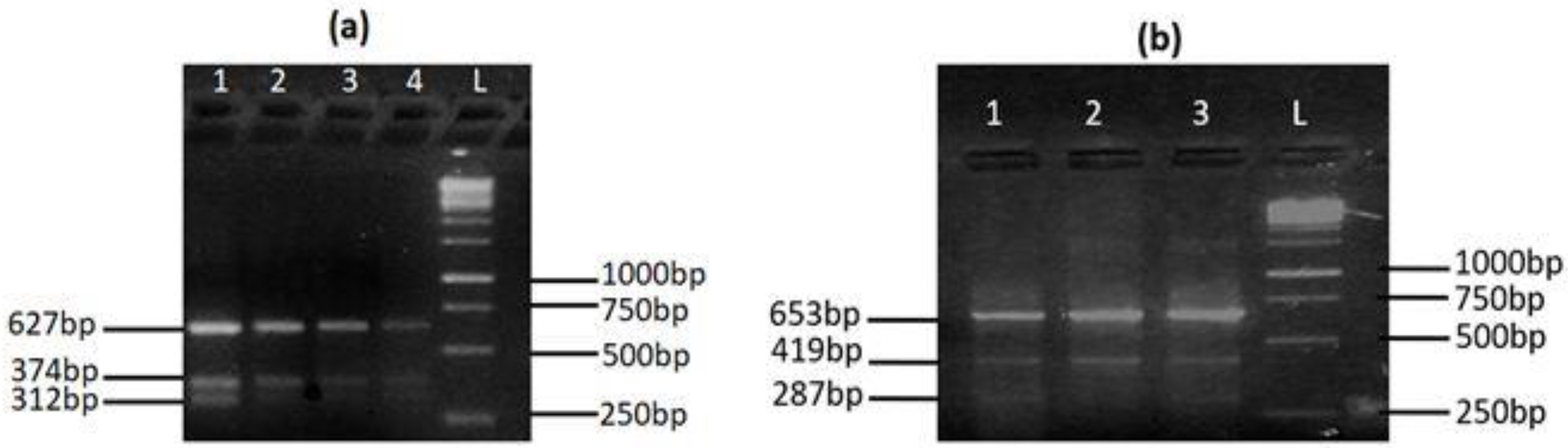
| MYH7rs121913645 | |||
| Forward outer | CAGCCGTGACCTCTCTGCATCAGAAGACAG | 64.7 | Control = 653 bp A allele = 287 bp G allele = 419 bp |
| Reverse outer | CTGAGCCTAGCAGATTCATGGCACTCACAGG | 64.6 | |
| Forward inner | GCACGCTTGAGGACCAGATCATCCCGA | 65.5 | |
| Reverse inner | GCCAAATGCCTCCAGAGCAGGGTTAGC | 65.2 | |
| Parameter | Control (n = 205) | Patient (n = 232) | Significance (p-Value) |
|---|---|---|---|
| Systolic BP (mmHg) | 118 ± 15 | 140 ± 12 | <0.001 |
| Diastolic BP (mmHg) | 79 ± 11 | 88 ± 15 | <0.001 |
| Ejection Fraction % (EF) | 55 ± 5 | 45 ± 10 | <0.001 |
| Creatine kinase (CK) (U/L) | 62 ± 41 | 149 ± 61 | <0.001 |
| CK-MB (ng/mL) | 3.7 ± 1.1 | 5.3 ± 2.4 | <0.001 |
| Cardiac Troponin I (cTnI) (ng/mL) | 0.02 ± 0.01 | 0.05 ± 0.02 | 0.067 |
| Cholesterol (mg/dL) | 185 ± 40 | 221 ± 50 | <0.001 |
| HDL (mg/dL) | 48 ± 7 | 40 ± 12 | 0.055 |
| LDL (mg/dL) | 83 ± 11 | 126 ± 40 | <0.001 |
| Triglycerides (mg/dL) | 275 ± 169 | 239 ± 159 | 0.048 |
| Serum Uric acid (mg/dL) | 5.9 ± 1.8 | 7.1 ± 2.7 | <0.001 |
| Urea (mg/dL) | 16 ± 10 | 38 ± 10 | <0.001 |
| Creatinine (mg/dL) | 0.7 ± 0.2 | 1.1 ± 0.8 | <0.001 |
| Variant | Control (n = 205) | Patient (n = 232) | Significance (p-Value) | |
|---|---|---|---|---|
| rs121913642 (T>C) | TT | 202 (98.5%) | 224 (96.6%) | χ2 = 12.35 p < 0.001 |
| TC | 3 (1.5%) | 8 (3.4%) | ||
| CC | 0 (0%) | 0 (0%) | ||
| T | 407 (99.3%) | 456 (98.2%) | χ2 = 10.40 p < 0.001 | |
| C | 3 (0.7%) | 8 (1.8%) | ||
| rs121913645 (G>A) | GG | 203 (99%) | 229 (98.7%) | χ2 = 0.141 p = 0.666 |
| GA | 2 (1%) | 3 (1.3%) | ||
| AA | 0 (0%) | 0 (0%) | ||
| G | 408 (99.5%) | 461 (99.3%) | χ2 = 0.106 p = 0.677 | |
| A | 2 (0.5%) | 3 (0.7%) | ||
| Parameter | MYH7 rs121913642 | MYH7 rs121913645 | ||||
|---|---|---|---|---|---|---|
| TT | TC | p-Value | GG | GA | p-Value | |
| Systolic BP (mmHg) | 141 ± 18 | 143 ± 18 | 0.605 | 142 ± 18 | 136 ± 18 | 0.142 |
| Diastolic BP (mmHg) | 80 ± 10 | 82 ± 10 | 0.346 | 82 ± 10 | 79 ± 9 | 0.210 |
| Ejection fraction (%) | 44 ± 10 | 39 ± 11 | 0.013 * | 43 ± 5 | 40 ± 11 | 0.001 * |
| Creatine kinase (CK) (U/L) | 148 ± 61 | 149 ± 60 | 0.930 | 145 ± 60 | 147 ± 60 | 0.873 |
| CK-MB (ng/mL) | 5.1 ± 2.4 | 5.5 ± 2.9 | 0.001 * | 5.3 ± 2.0 | 5.3 ± 2.4 | 0.763 |
| Cardiac troponin I (cTnI) (ng/mL) | 0.05 ± 0.02 | 0.06 ± 0.03 | 0.007 * | 0.05 ± 0.02 | 0.05 ± 0.01 | 0.757 |
| Cholesterol (mg/dL) | 226 ± 50 | 212 ± 45 | 0.095 | 222 ± 60 | 212 ± 45 | 0.082 |
| HDL Cholesterol (mg/dL) | 64 ± 58 | 53 ± 18 | 0.161 | 60 ± 40 | 71 ± 50 | 0.474 |
| LDL cholesterol (mg/dL) | 132 ± 80 | 112 ± 57 | 0.104 | 122 ± 58 | 148 ± 130 | 0.011 * |
| Triglycerides (mg/dL) | 245 ± 164 | 229 ± 119 | 0.561 | 238 ± 144 | 254 ± 187 | 0.997 |
| Urea (mg/dL) | 43 ± 19 | 41 ± 20 | 0.566 | 41 ± 19 | 46 ± 18 | 0.869 |
| Uric acid (mg/dL) | 7.1 ± 2.7 | 7.1 ± 2.5 | 0.882 | 7.1 ± 2.6 | 7.1 ± 3.1 | 0.221 |
| Creatinine(mg/dL) | 1.1 ± 0.6 | 1.1 ± 0.5 | 0.978 | 1.0 ± 0.4 | 1.3 ± 0.6 | 0.046 * |
| Variant | Allele | Allele | Variant | Allele | Allele |
|---|---|---|---|---|---|
| rs121913642 | T | C | rs121913645 | G | A |
| Ref. | 1.982 (1.317–2.982) p < 0.001 * | Ref. | 0.878 (0.506–1.522) p = 0.642 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousaf, M.; Khan, W.A.; Shahzad, K.; Khan, H.N.; Ali, B.; Hussain, M.; Awan, F.R.; Mustafa, H.; Sheikh, F.N. Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction. Genes 2022, 13, 1554. https://doi.org/10.3390/genes13091554
Yousaf M, Khan WA, Shahzad K, Khan HN, Ali B, Hussain M, Awan FR, Mustafa H, Sheikh FN. Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction. Genes. 2022; 13(9):1554. https://doi.org/10.3390/genes13091554
Chicago/Turabian StyleYousaf, Memoona, Waqas Ahmed Khan, Khurrum Shahzad, Haq Nawaz Khan, Basharat Ali, Misbah Hussain, Fazli Rabbi Awan, Hamid Mustafa, and Farah Nadia Sheikh. 2022. "Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction" Genes 13, no. 9: 1554. https://doi.org/10.3390/genes13091554
APA StyleYousaf, M., Khan, W. A., Shahzad, K., Khan, H. N., Ali, B., Hussain, M., Awan, F. R., Mustafa, H., & Sheikh, F. N. (2022). Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction. Genes, 13(9), 1554. https://doi.org/10.3390/genes13091554