

Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Sample and Data Collection
2.3. Next-Generation Sequencing
2.4. Statistical Analysis
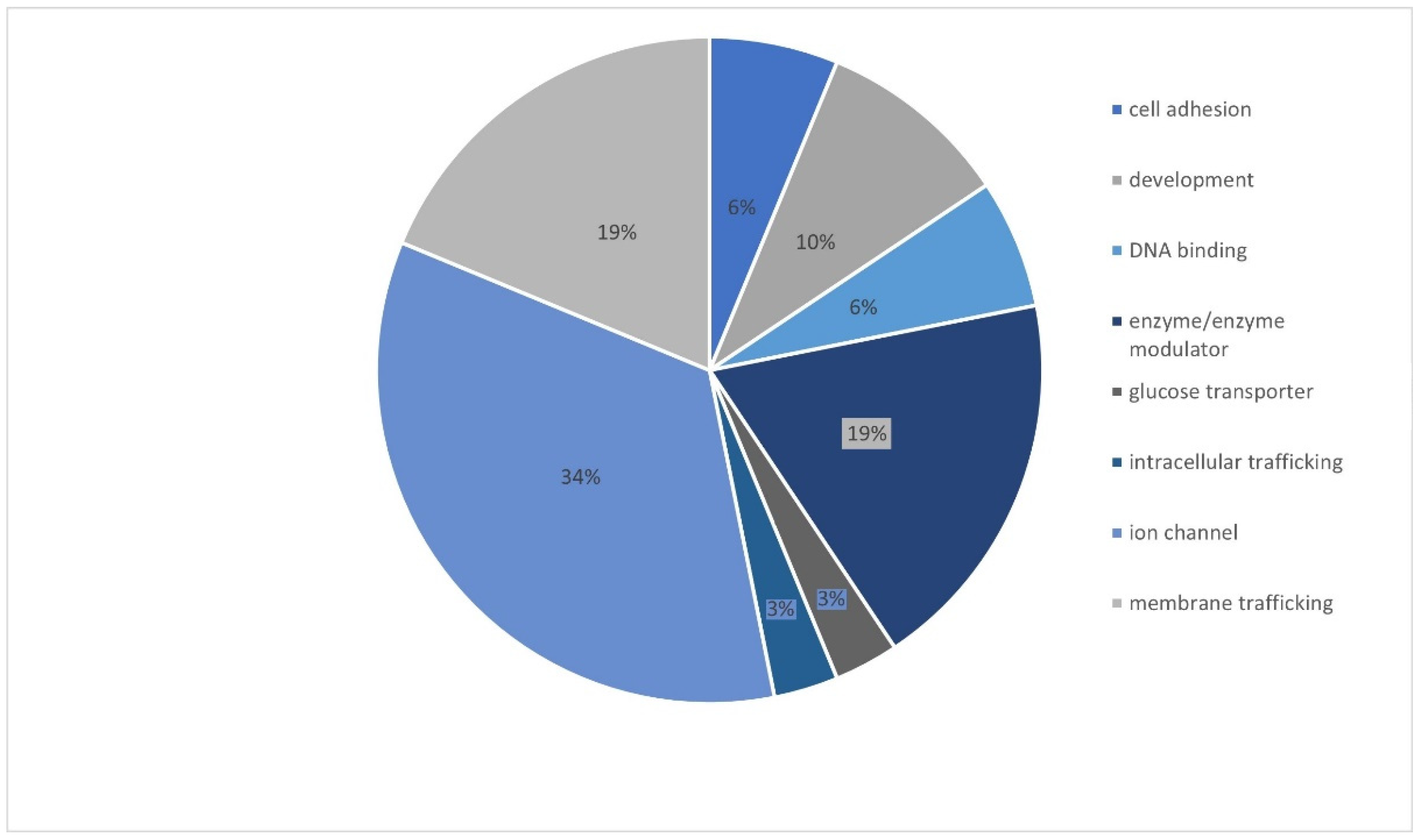
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scala, M.; Bianchi, A.; Bisulli, F.; Coppola, A.; Elia, M.; Trivisano, M.; Pruna, D.; Pippucci, T.; Canafoglia, L.; Lattanzi, S.; et al. Advances in genetic testing and optimization of clinical management in children and adults with epilepsy. Expert Rev. Neurother. 2020, 20, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Zuberi, S.M.; Johnson, M.R. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 2017, 30, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Falco-Walter, J.J.; Scheffer, I.E.; Fisher, R.S. The new definition and classification of seizures and epilepsy. Epilepsy Res. 2018, 139, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Annegers, J.F.; Hauser, W.A.; Anderson, V.E.; Kurland, L.T. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology 1982, 32, 174. [Google Scholar] [CrossRef]
- Myers, C.T.; Mefford, H.C. Advancing epilepsy genetics in the genomic era. Genome Med. 2015, 7, 91. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Salinas, V.; Martínez, N.; Maturo, J.P.; Rodriguez-Quiroga, S.A.; Zavala, L.; Medina, N.; Amartino, H.; Sfaello, I.; Agosta, G.; Serafín, E.M.; et al. Clinical next generation sequencing in developmental and epileptic encephalopathies: Diagnostic relevance of data re-analysis and variants re-interpretation. Eur. J. Med. Genet. 2021, 64, 104363. [Google Scholar] [CrossRef]
- Miao, P.; Feng, J.; Guo, Y.; Wang, J.; Xu, X.; Wang, Y.; Li, Y.; Gao, L.; Zheng, C.; Cheng, H. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin. Genet. 2018, 94, 512–520. [Google Scholar] [CrossRef]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef]
- Hoelz, H.; Herdl, C.; Gerstl, L.; Tacke, M.; Vill, K.; von Stuelpnagel, C.; Rost, I.; Hoertnagel, K.; Abicht, A.; Hollizeck, S.; et al. Impact on Clinical Decision Making of Next-Generation Sequencing in Pediatric Epilepsy in a Tertiary Epilepsy Referral Center. Clin. EEG Neurosci. 2020, 51, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdò, S.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes: HUMAN MUTATION. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Moreno, L.; Giráldez, B.G.; Soto-Insuga, V.; Losada-Del Pozo, R.; Rodrigo-Moreno, M.; Alarcón-Morcillo, C.; Sánchez-Martín, G.; Díaz-Gómez, E.; Guerrero-López, R.; Serratosa, J.M.; et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS ONE 2017, 12, e0188978. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Cordeiro, D.; Hewson, S.; Cohn, R.; Kannu, P.; Kobayashi, J.; Mahmutoglu, S. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. J. Neurol. Sci. 2015, 357, e31. [Google Scholar] [CrossRef]
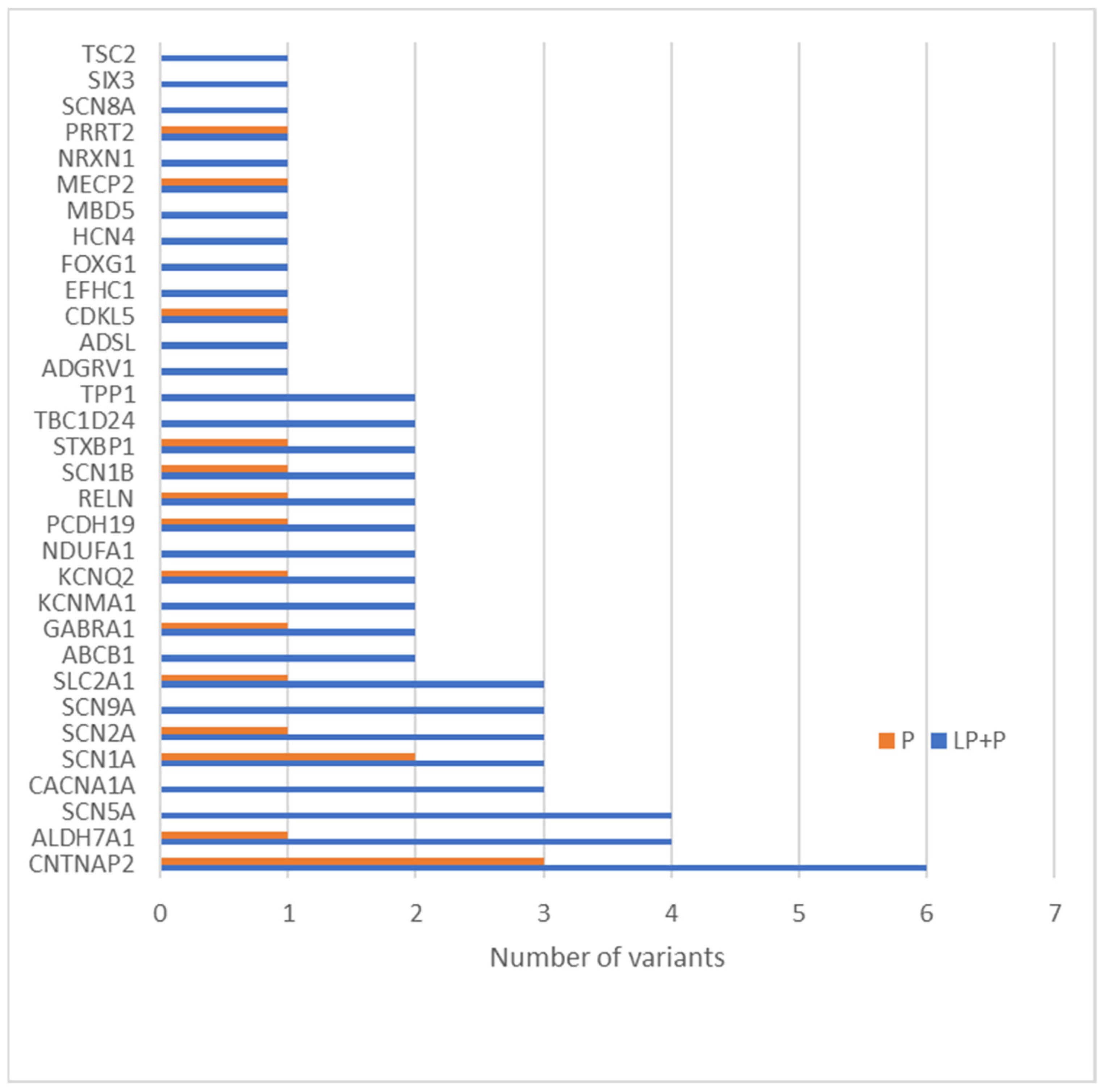
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Saint-Martin, M.; Joubert, B.; Pellier-Monnin, V.; Pascual, O.; Noraz, N.; Honnorat, J. Contactin-associated protein-like 2, a protein of the neurexin family involved in several human diseases. Eur. J. Neurosci. 2018, 48, 1906–1923. [Google Scholar] [CrossRef]
- Strauss, K.A.; Puffenberger, E.G.; Huentelman, M.J.; Gottlieb, S.; Dobrin, S.E.; Parod, J.M.; Stephan, D.A.; Morton, D.H. Recessive Symptomatic Focal Epilepsy and Mutant Contactin-Associated Protein-like 2. N. Engl. J. Med. 2006, 354, 1370–1377. [Google Scholar] [CrossRef]
- Mutational analysis of the SCN1A, SCN1B and GABRG2 genes in 150 Italian patients with idiopathic childhood epilepsies. Clin. Genet. 2009, 75, 579–581. [CrossRef]
- Carranza Rojo, D.; Hamiwka, L.; McMahon, J.M.; Dibbens, L.M.; Arsov, T.; Suls, A.; Stodberg, T.; Kelley, K.; Wirrell, E.; Appleton, B.; et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology 2011, 77, 380–383. [Google Scholar] [CrossRef]
- Sadleir, L.G.; Mountier, E.I.; Gill, D.; Davis, S.; Joshi, C.; DeVile, C.; Kurian, M.A.; For the DDD Study; Mandelstam, S.; Wirrell, E.; et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology 2017, 89, 1035–1042. [Google Scholar] [CrossRef]
- Ogiwara, I.; Ito, K.; Sawaishi, Y.; Osaka, H.; Mazaki, E.; Inoue, I.; Montal, M.; Hashikawa, T.; Shike, T.; Fujiwara, T.; et al. De novo mutations of voltage-gated sodium channel II gene SCN2A in intractable epilepsies. Neurology 2009, 73, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Hahn, A.; Bast, T.; Müller, S.; Löffler, H.; Maljevic, S.; Gaily, E.; Prehl, I.; Biskup, S.; Joensuu, T.; et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 2016, 263, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, S.F.; Heron, S.E.; Giordano, L.; Marini, C.; Guerrini, R.; Kaplan, R.E.; Gambardella, A.; Steinlein, O.K.; Grinton, B.E.; Dean, J.T.; et al. Benign familial neonatal-infantile seizures: Characterization of a new sodium channelopathy. Ann. Neurol. 2004, 55, 550–557. [Google Scholar] [CrossRef]
- Kato, M.; Yamagata, T.; Kubota, M.; Arai, H.; Yamashita, S.; Nakagawa, T.; FujII, T.; Sugai, K.; Imai, K.; Uster, T.; et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013, 54, 1282–1287. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, E.C.; Chen, C.; Procko, E.; Pant, S.; Lam, K.; Patel, J.; Choi, R.; Hong, M.; Joshi, D.; et al. Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy. Sci. Rep. 2020, 10, 4756. [Google Scholar] [CrossRef]
- Horák, O.; Burešová, M.; Kolář, S.; Španělová, K.; Jeřábková, B.; Gaillyová, R.; Česká, K.; Réblová, K.; Šoukalová, J.; Zídková, J.; et al. Next-generation sequencing in children with epilepsy: The importance of precise genotype–phenotype correlation. Epilepsy Behav. 2022, 128, 108564. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Cohort (n = 277) | VUS 1 (n = 54) | LP 1 (n = 47) | P 1 (n = 17) | |
| Male/female (%) | 52/48 | 61.1/38.9 | 42.6/57.4 | 47.1/52.9 |
| Mean age at sample sequencing (range) | 8.4 (0.5–17.5) | 8.8 (1–17.5) | 7.7 (0.5–17) | 7 (1–14) |
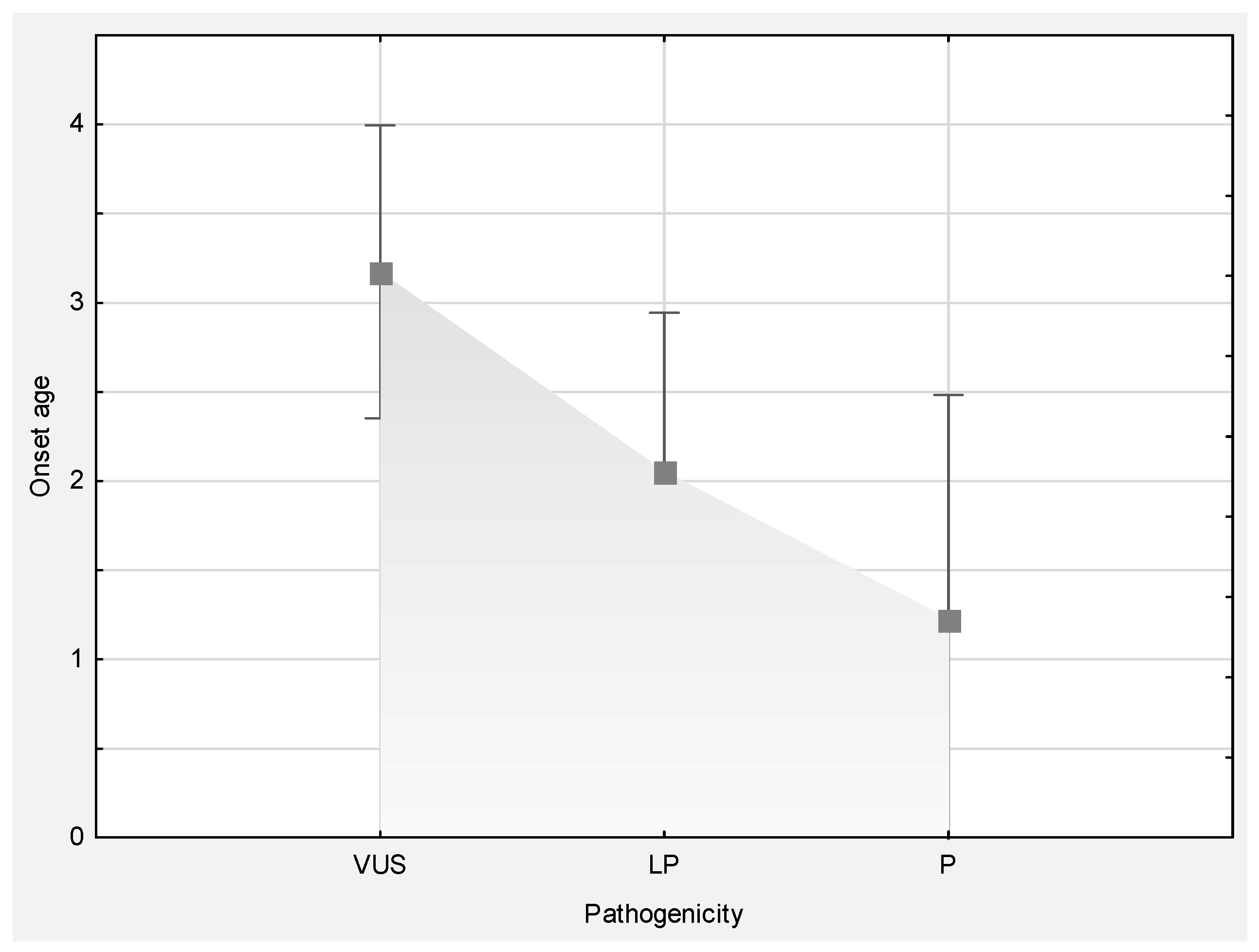
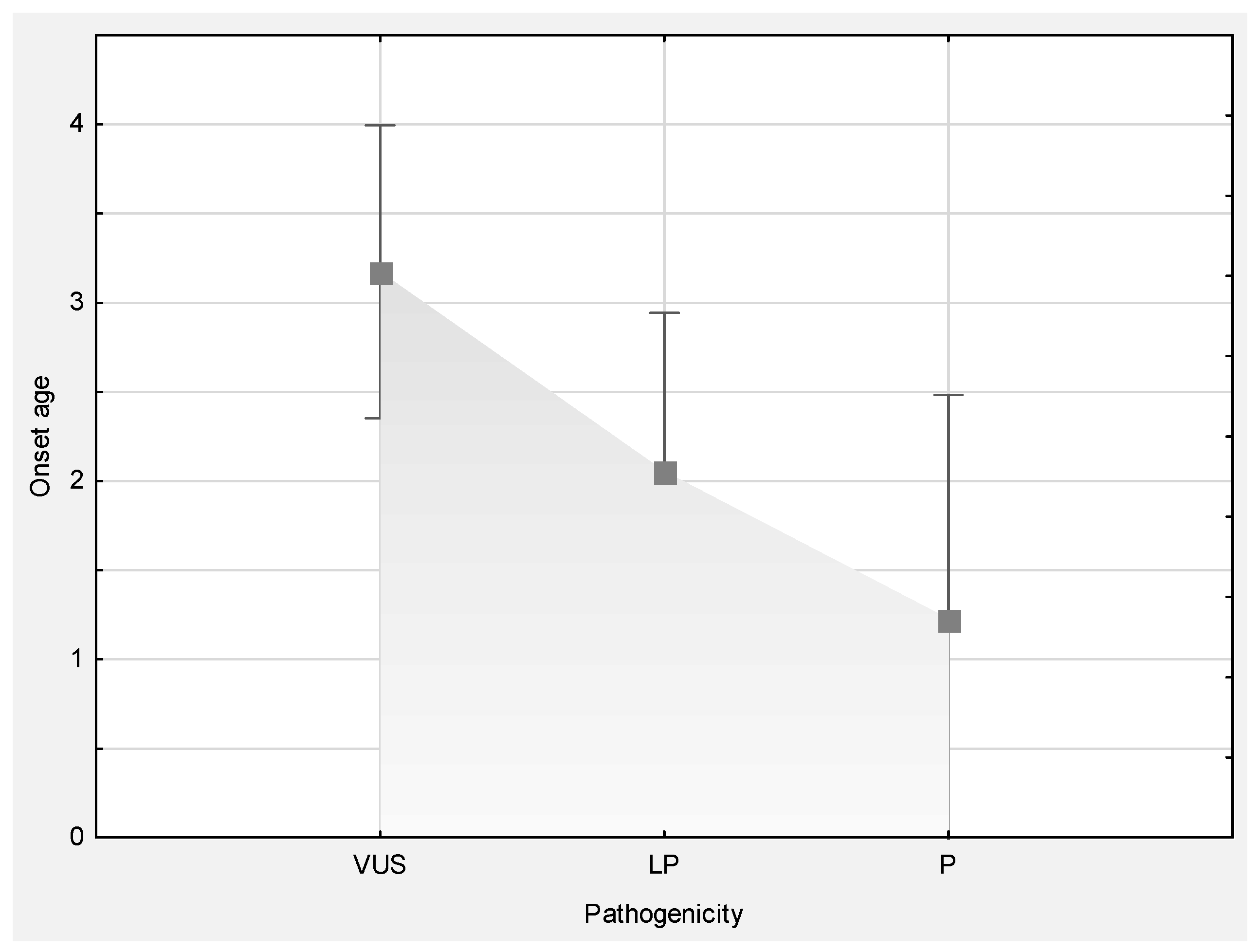
| Mean age at disease onset (range) | 3 (0–15.5) | 3.2 (0–12) | 2.1 (0–10.5) | 1.2 (0–4) |
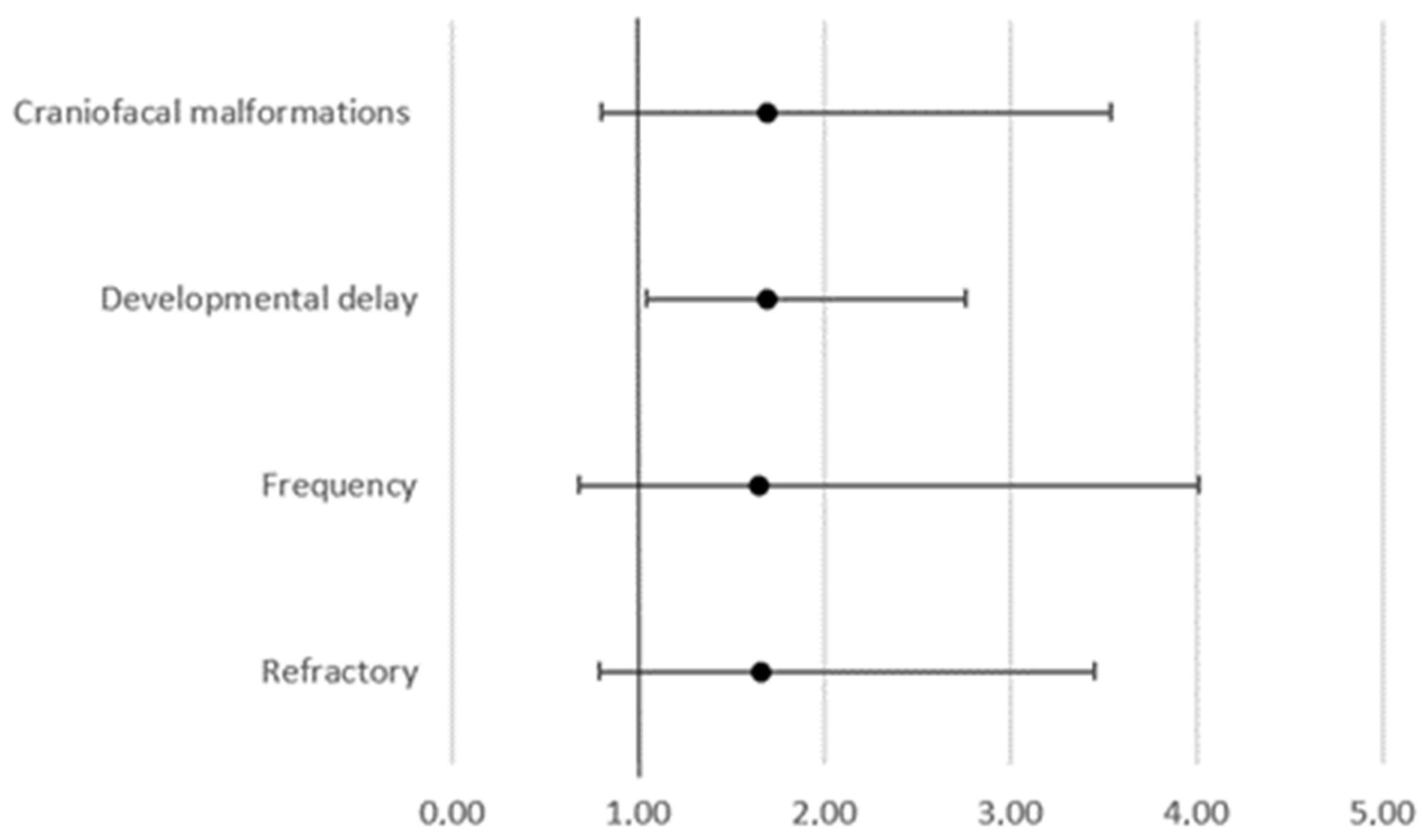
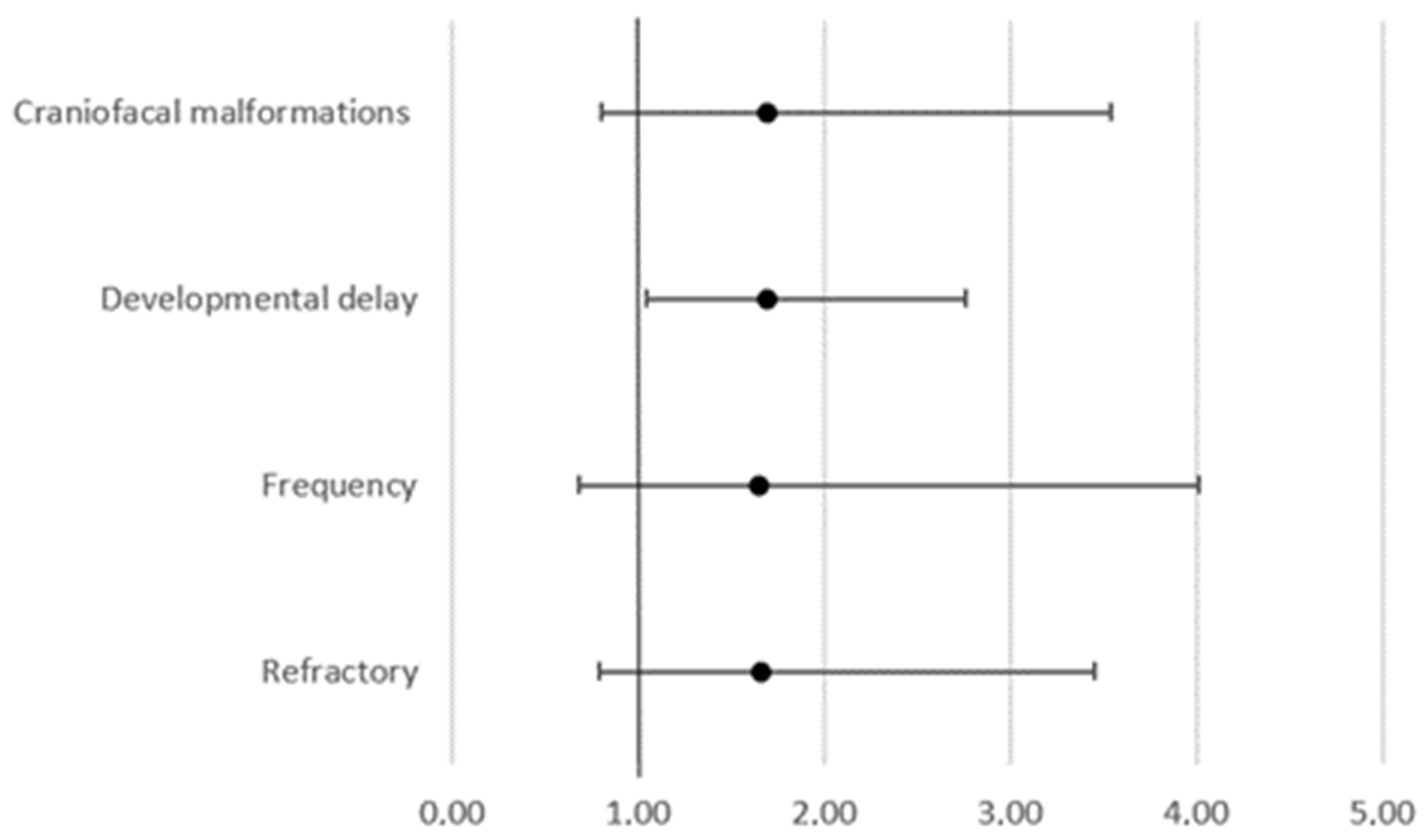
| Focal epilepsy | 14.1% | 9.3% | 12.8% | 11.8% |
| Generalized epilepsy | 41.2% | 40.7% | 57.4% | 47.1% |
| Combined epilepsy | 34.3% | 37.0% | 23.4% | 35.3% |
| Syndrome | 16.6% | 13.0% | 19.1% | 29.4% |
| Refractory | 51.6% | 55.6% | 57.4% | 17.6% |
| Speech delay | 13.7% | 9.3% | 27.7% | 52.9% |
| Learning disability | 10.1% | 11.1% | 17.0% | 52.9% |
| Behavioral changes | 6.9% | 13.0% | 4.3% | 5.9% |
| Psychomotor retardation | 27.4% | 25.9% | 38.3% | 0.0% |
| Craniofacial malformations | 11.6% | 18.5% | 14.9% | 0.0% |
| Abnormal EEG | 72.6% | 83.3% | 55.3% | 41.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blazekovic, A.; Gotovac Jercic, K.; Meglaj, S.; Duranovic, V.; Prpic, I.; Lozic, B.; Malenica, M.; Markovic, S.; Lujic, L.; Petelin Gadze, Z.; et al. Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice. Genes 2022, 13, 1466. https://doi.org/10.3390/genes13081466
Blazekovic A, Gotovac Jercic K, Meglaj S, Duranovic V, Prpic I, Lozic B, Malenica M, Markovic S, Lujic L, Petelin Gadze Z, et al. Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice. Genes. 2022; 13(8):1466. https://doi.org/10.3390/genes13081466
Chicago/Turabian StyleBlazekovic, Antonela, Kristina Gotovac Jercic, Sarah Meglaj, Vlasta Duranovic, Igor Prpic, Bernarda Lozic, Masa Malenica, Silvana Markovic, Lucija Lujic, Zeljka Petelin Gadze, and et al. 2022. "Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice" Genes 13, no. 8: 1466. https://doi.org/10.3390/genes13081466
APA StyleBlazekovic, A., Gotovac Jercic, K., Meglaj, S., Duranovic, V., Prpic, I., Lozic, B., Malenica, M., Markovic, S., Lujic, L., Petelin Gadze, Z., Juraski, R. G., Barišic, N., Baric, I., & Borovecki, F. (2022). Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice. Genes, 13(8), 1466. https://doi.org/10.3390/genes13081466