
Identification of the Key Pathways and Genes Involved in the Wax Biosynthesis of the Chinese White Wax Scale Insect (Ericerus pela Chavannes) by Integrated Weighted Gene Coexpression Network Analysis

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. High-Throughput Transcriptome Sequencing
2.3. Weighted Correlation Network Analysis
2.3.1. Data Input and Preprocessing
2.3.2. Gene Network Construction and Module Identification
2.3.3. Relating Modules with Wax Secretion Trait Data
2.3.4. Network Visualization
2.4. Selection of Hub Genes
2.5. Drawing of Figures
3. Results
3.1. Transcriptome Sequencing Results
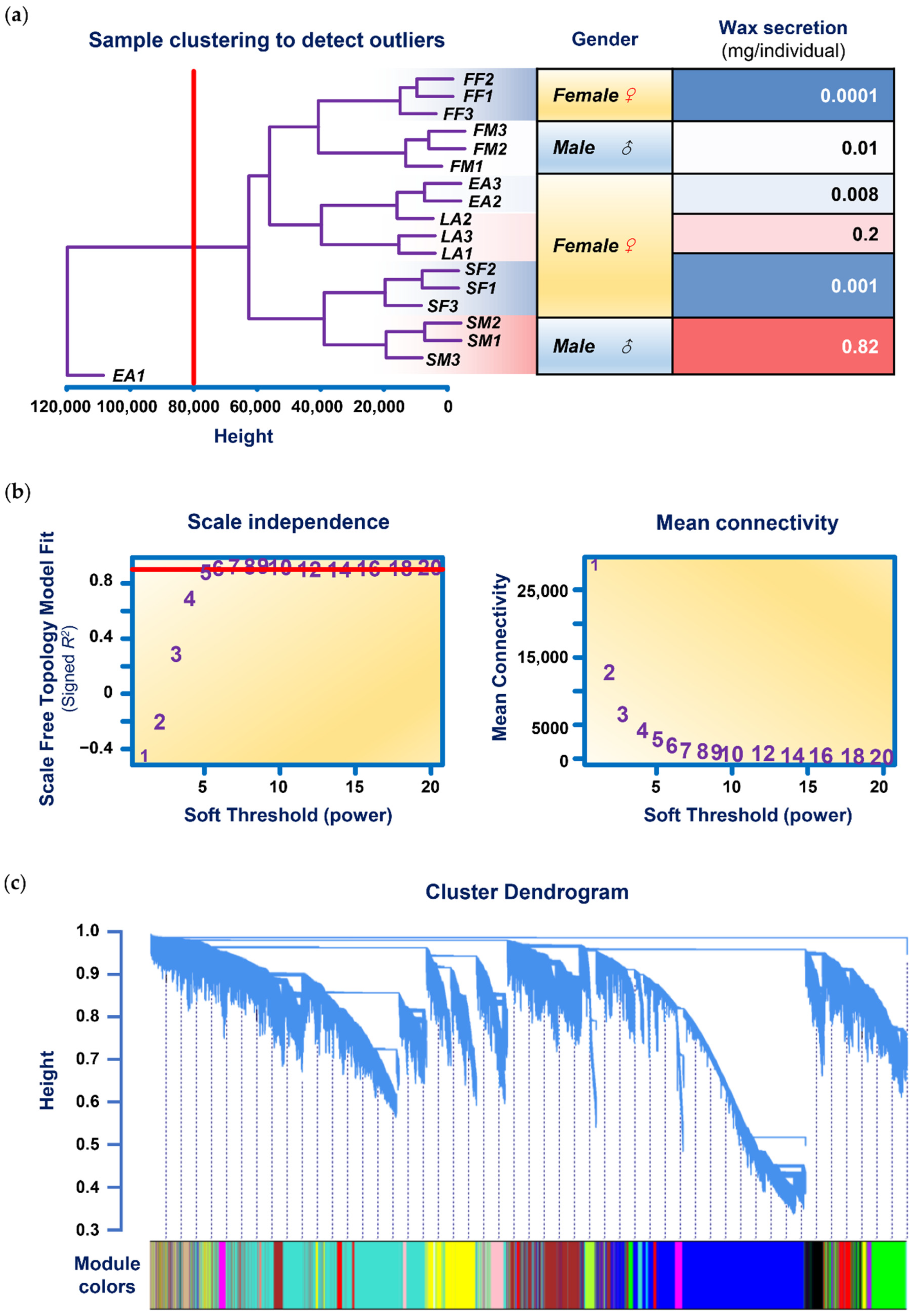
3.2. Data Input, Cleaning, and Pre-Processing

3.3. Analysis of Network Topology
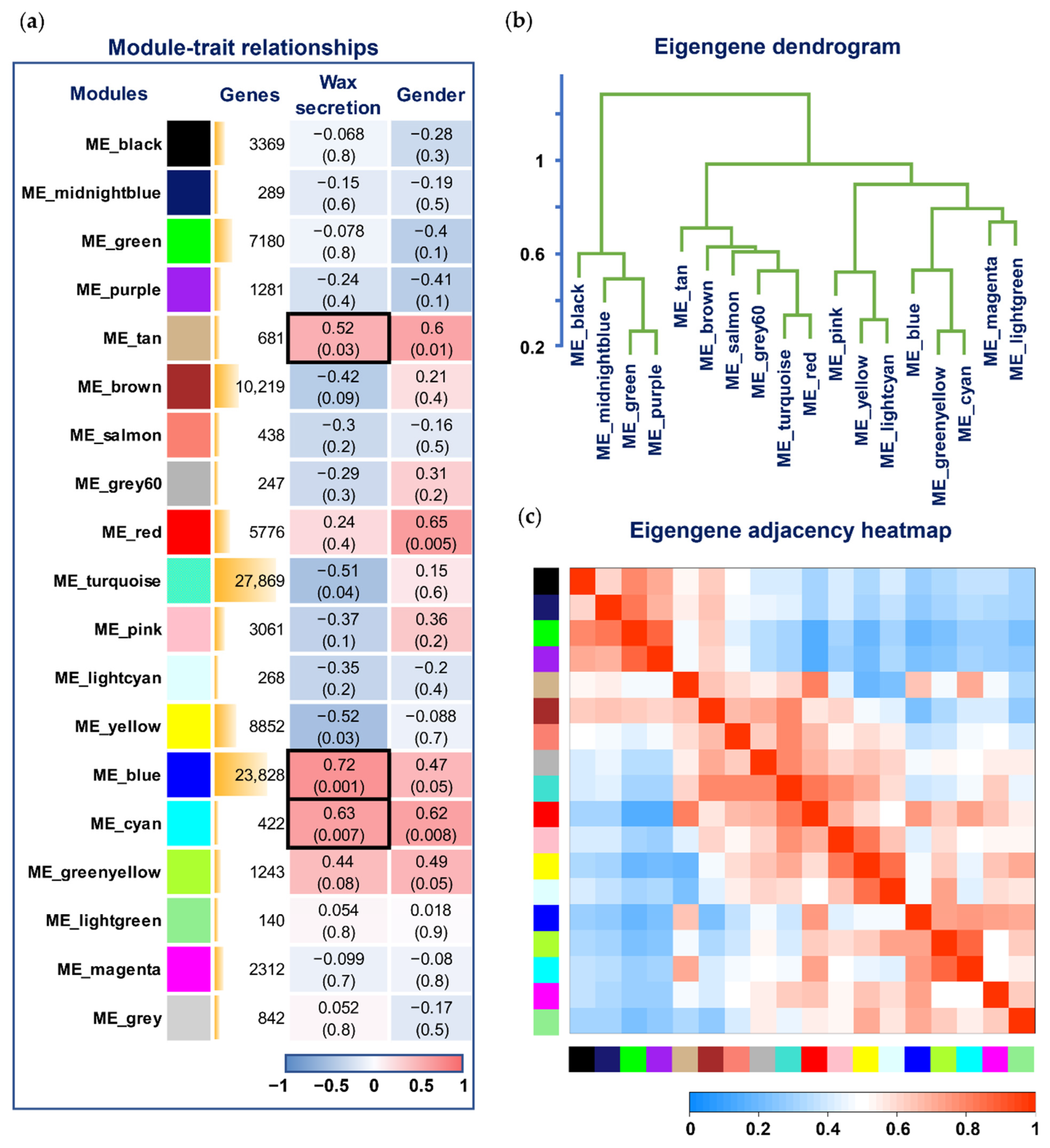
3.4. Network Construction and Module Detection


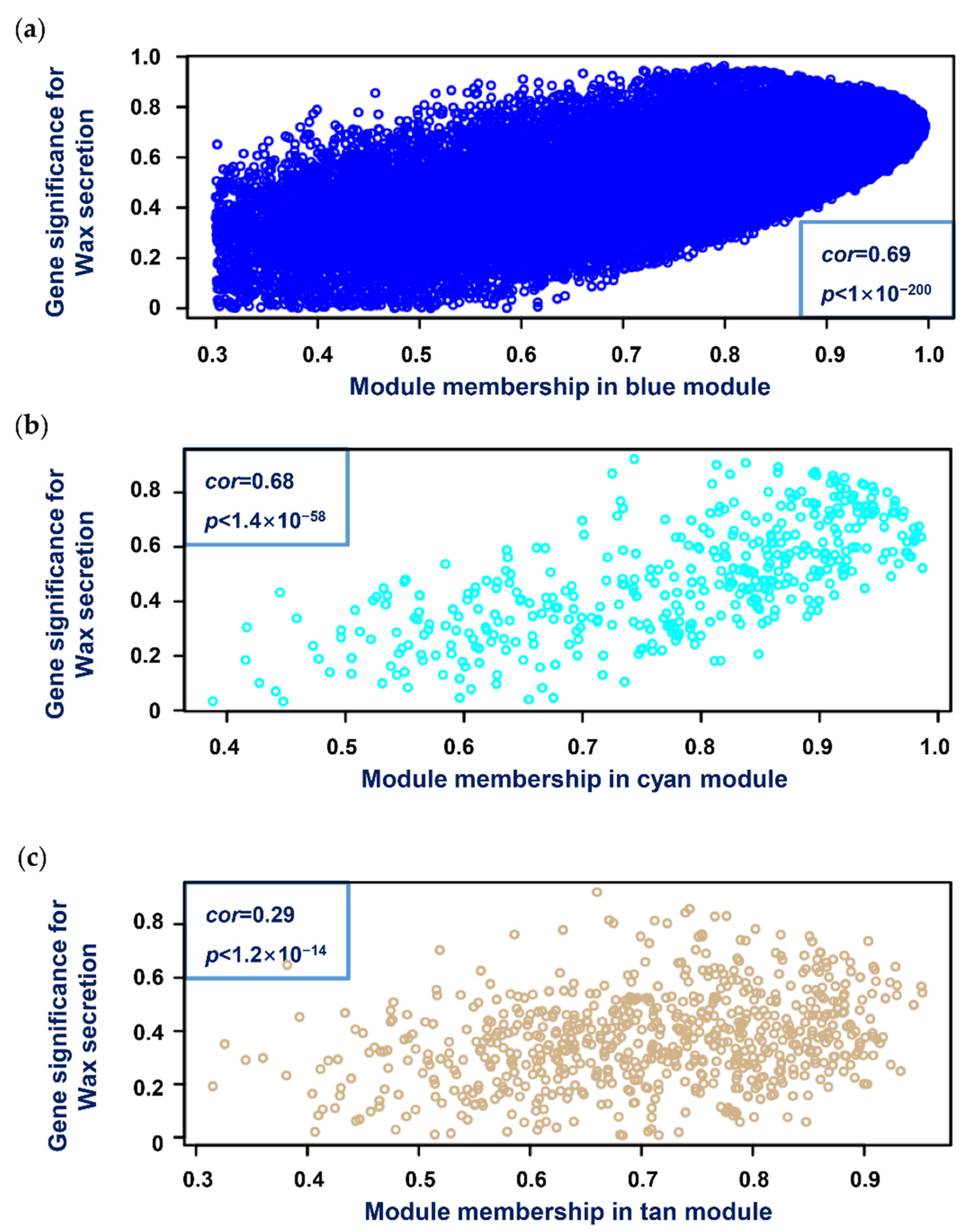
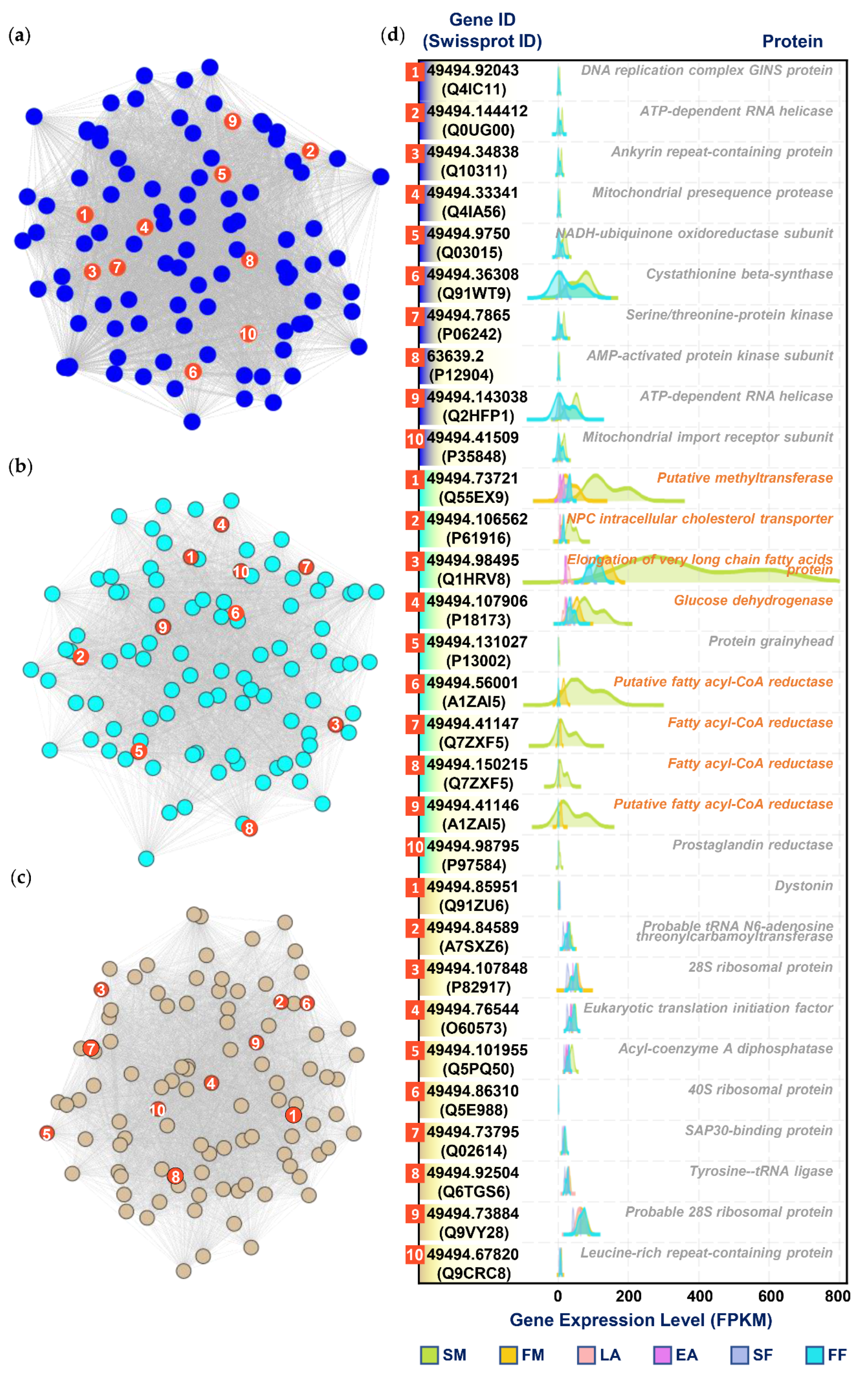
3.5. Selection of Hub Genes


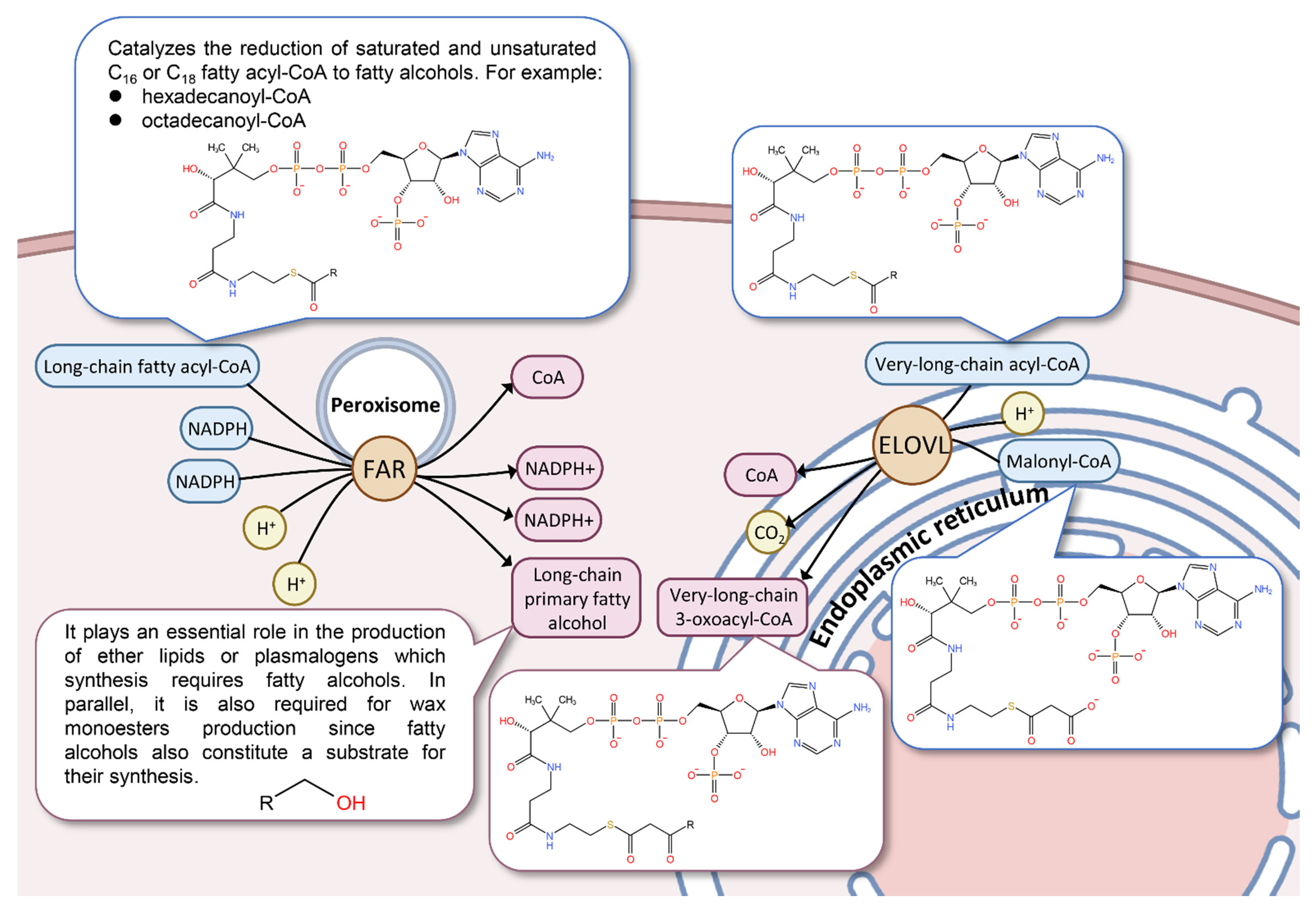
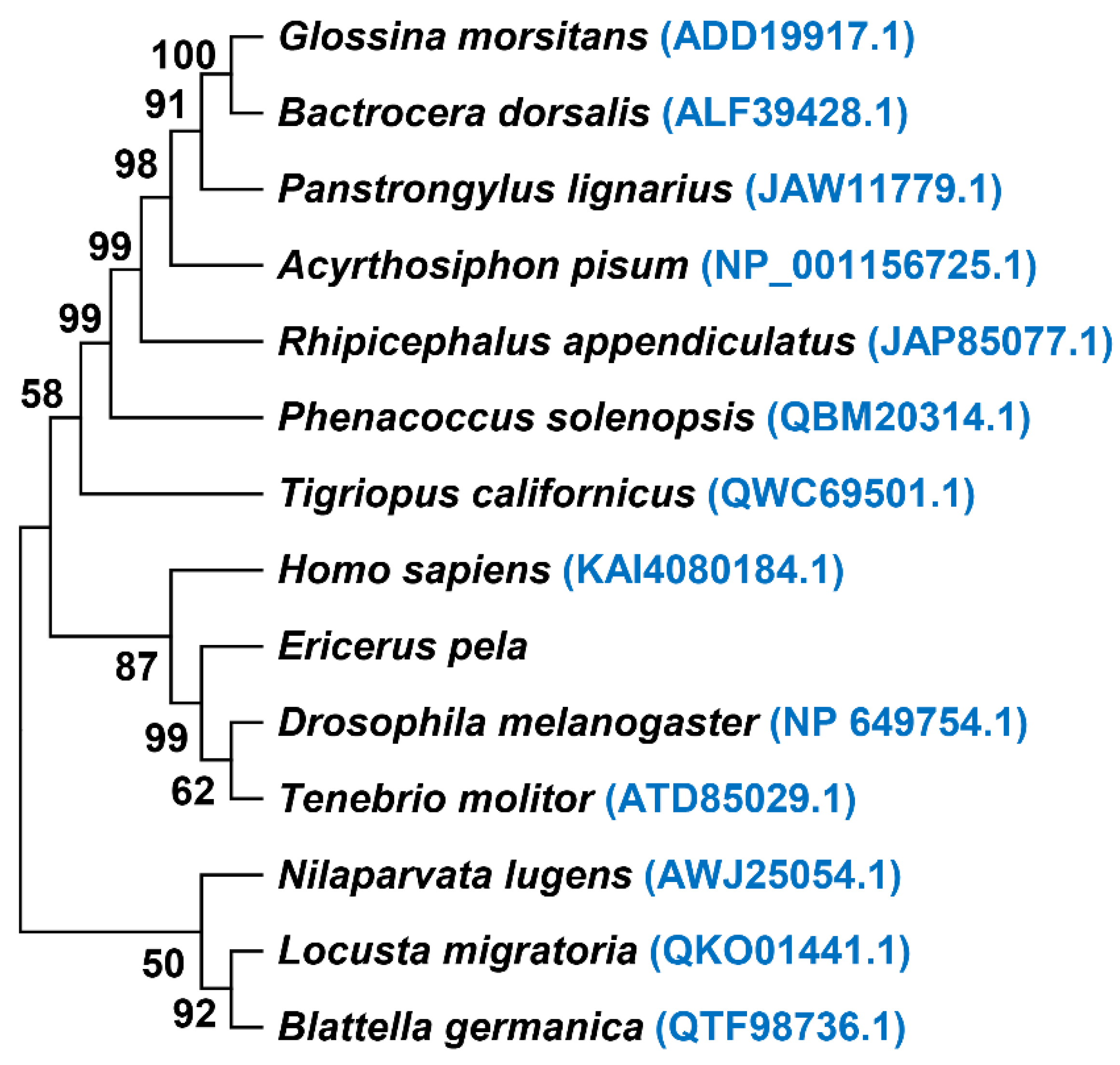
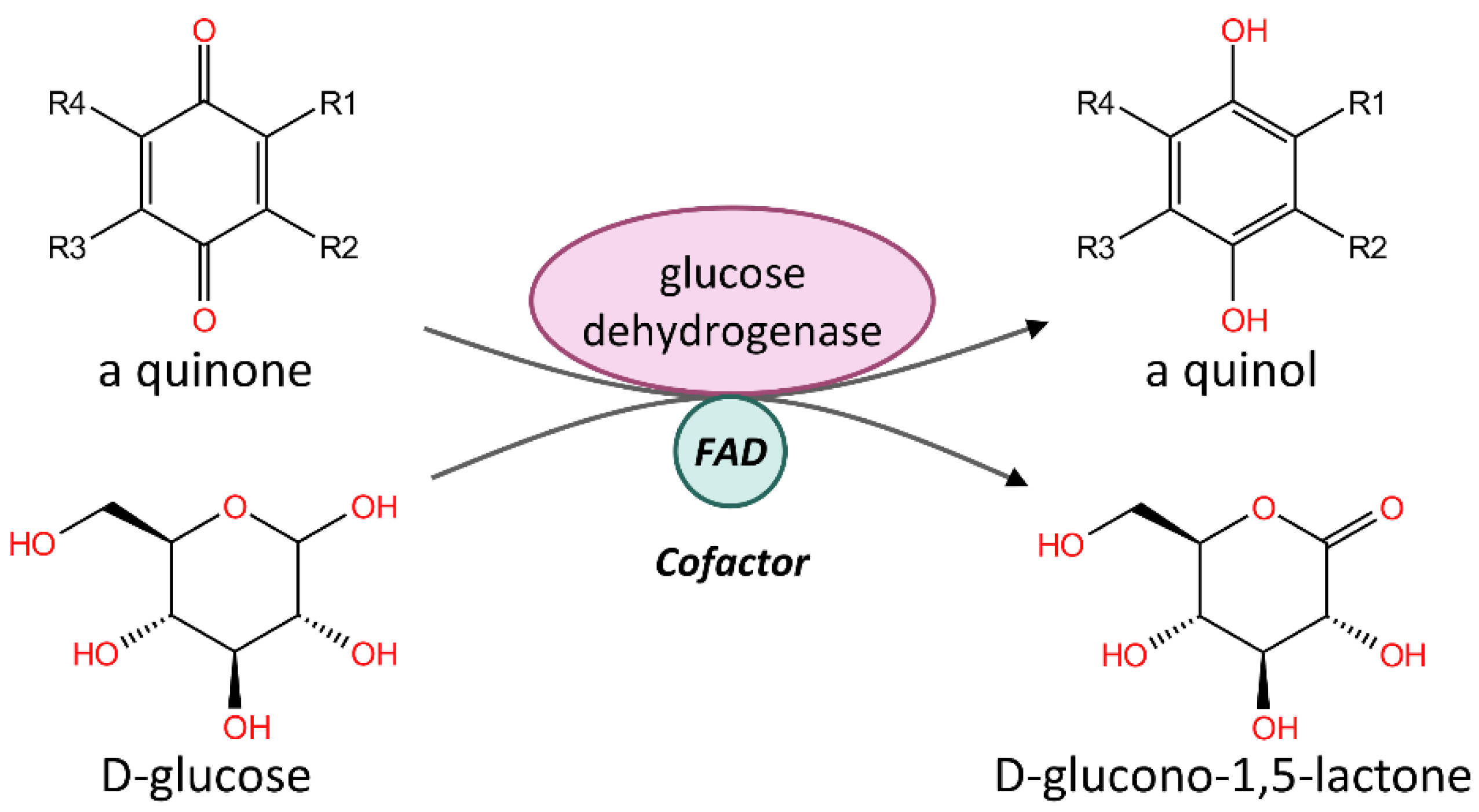
4. Discussion



5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.M.; Wang, Z.L.; Chen, Y.; Wang, S.Y.; Feng, Y. The Impact of Enviromental Factors on the Wax Excretion by Chinese White Wax Scale (Ericerus pela Chavannes). Acta Ecol. Sin. 2007, 27, 103–112. [Google Scholar]
- Chen, X.M.; Wang, Z.L.; Chen, Y.; Zhao, J.J.; Ye, S.D.; Wang, S.Y. Design and Analysis of a System for Breeding White Wax Scale Insects and Producing Insect Wax in the Same Region. Sci. Silvae Sin. 2010, 46, 106–113. [Google Scholar]
- Liu, N.; Lu, Q.; Zhang, J.W.; Ling, X.F.; Liu, H.P.; Chen, H.; Chen, X.M. Impact of Exogenous Juvenile Hormone Analogue on Wax Excretion and Development of Chinese White Wax Scale (Ericerus pela Chavannes). For. Res. 2018, 31, 114–119. [Google Scholar] [CrossRef]
- Chen, H.; Lu, Q.; Chen, X.M.; Ling, X.F.; Liu, P.F.; Liu, N.; Wang, W.W.; Zhang, J.W.; Qi, Q.; Ding, W.F.; et al. Genome Assembly and Methylome Analysis of the White Wax Scale Insect Provides Insight into Sexual Differentiation of Metamorphosis in Hexapods. Mol. Ecol. Resour. 2021, 21, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Chen, X.M.; Liu, W.W.; Feng, Y.; Sun, T. Transcriptome Analysis of Sexually Dimorphic Chinese White Wax Scale Insects Reveals Key Differences in Developmental Programs and Transcription Factor Expression. Sci. Rep. 2015, 5, 8141. [Google Scholar] [CrossRef]
- Chen, X.M.; Wang, Z.L.; Chen, Y.; Wang, S.Y.; Feng, Y. The Main Climate Factors Affecting Wax Excretion of Ericerus pela Chavannes (Homopetera: Coccidae) and an Analysis of Its Ecological Adaptability. Acta Entomol. Sin. 2007, 50, 136–143. [Google Scholar]
- Zhao, Z.L.; Yu, S.H.; Zhou, W.W.; Zhu, Z.R.; Guo, Z.J.; Yang, P. The Composition and Function of the Honeydew Secreted by Ericerus pela as Deduced from Metabolome and Transcriptome Data. Chin. J. Appl. Entomol. 2019, 56, 1007–1018. [Google Scholar] [CrossRef]
- Millar, A.A.; Clemens, S.; Zachgo, S.; Giblin, E.M.; Taylor, D.C.; Kunst, L. CUT1, an Arabidopsis Gene Required for Cuticular Wax Biosynthesis and Pollen Fertility, Encodes a Very-Long-Chain Fatty Acid Condensing Enzyme. Plant Cell 1999, 11, 825–838. [Google Scholar] [CrossRef]
- Yang, P.; Xu, D.L.; Chen, X.M.; Liu, W.W.; Gong, Z.J.; Hu, Y.H. Research Progress on Wax Ester Biosynthetic Pathway and the Related Key Enzymes. Chin. J. Cell Biol. 2012, 34, 695–703. [Google Scholar]
- Kunst, L.; Samuels, A.L. Biosynthesis and Secretion of Plant Cuticular Wax. Prog. Lipid Res. 2003, 42, 51–80. [Google Scholar] [CrossRef]
- Klypina, N.; Hanson, S.F. Arabidopsis thaliana Wax Synthase Gene Homologues Show Diverse Expression Patterns that Suggest a Specialized Role for These Genes in Reproductive Organs. Plant Sci. 2008, 175, 312–320. [Google Scholar] [CrossRef]
- Doan, T.T.P.; Carlsson, A.S.; Hamberg, M.; Bülow, L.; Stymne, S.; Olsson, P. Functional expression of five Arabidopsis fatty acyl-CoA reductase genes in Escherichia coli. J. Plant Physiol. 2009, 166, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.W.; Wang, X.Q.; Sun, T.; Qi, Q.; Yu, S.H.; Yang, P.; Chen, X.M. Cloning and Prokaryotic Expression of Wax Synthase Gene of the Chinese White Wax Scale. For. Res. 2016, 29, 610–614. [Google Scholar] [CrossRef]
- Cheng, J.B.; Russell, D.W. Mammalian Wax Biosynthesis II. Expression Cloning of Wax Synthase cDNAs Encoding a Member of the Acyltransferase Enzyme Family. J. Biol. Chem. 2004, 279, 37798–37807. [Google Scholar]
- Cheng, J.B.; Russell, D.W. Mammalian Wax Biosynthesis, I. Identification of Two Fatty Acyl-coenzyme a Reductases with Different Substrate Specificities and Tissue Distributions. J. Biol. Chem. 2004, 279, 37789–37797. [Google Scholar] [CrossRef]
- Yang, P.; Zhu, J.Y.; Gong, Z.J.; Xu, D.L.; Chen, X.M.; Liu, W.W.; Lin, X.D.; Li, Y.F. Transcriptome Analysis of the Chinese White Wax Scale Ericerus pela with Focus on Genes Involved in Wax Biosynthesis. PLoS ONE 2012, 7, e35719. [Google Scholar] [CrossRef][Green Version]
- Hu, Y.H.; Chen, X.M.; Yang, P.; Ding, W.F. Characterization and Functional Assay of a Fatty Acyl-CoA Reductase Gene in the Scale Insect, Ericerus pela Chavannes (Hemiptera: Coccoidae). Arch. Insect Biochem. Physiol. 2017, 97, e21445. [Google Scholar] [CrossRef]
- Zhao, Z.L.; Wu, G.X.; GAO, X.; Yu, S.H.; Yang, B.; Zhu, J.Y.; Yang, P. Full-length Transcriptome Sequencing of Ericerus pela at the Early Stage of Wax Secretion and Protein Expression Analysis of FAR in Males and Females. J. Yunnan Agric. Univ. 2019, 34, 762–771. [Google Scholar] [CrossRef]
- Qi, Q.; Lv, P.; Chen, X.M.; Chen, H.; Chen, M.S.; Yang, P. Sexual Dimorphism in Wax Secretion Offers Ecological Adaptability During Ericerus pela (Hemiptera: Coccidae) Evolution. Environ. Entomol. 2019, 48, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Wang, W.W.; Ling, X.F.; Lu, Q.; Zhang, J.W.; He, R.; Chen, H. Regulation of Hormone-Related Genes in Ericerus pela (Hemiptera: Coccidae) for Dimorphic Metamorphosis. J. Insect Sci. 2019, 19, 16. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Tutorials for the WGCNA Package. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/index.html (accessed on 15 June 2022).
- Shannon, P.; Markie, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.X.; Pan, Y.; Wu, F.X. CytoNCA: A Cytoscape Plugin for Centrality Analysis and Evaluation of Protein Interaction Networks. BioSystems 2015, 127, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling Differential Gene Expression Analysis for De novo Assembled Transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; Martin, M.J.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, 115–119. [Google Scholar] [CrossRef]
- R: Distance Matrix Computation. Available online: https://stat.ethz.ch/R-manual/R-devel/library/stats/html/dist.html (accessed on 20 July 2022).
- Hierarchical Clustering-Wikipedia. Available online: https://en.wikipedia.org/wiki/Hierarchical_clustering (accessed on 20 July 2022).
- R: Hierarchical Clustering. Available online: https://stat.ethz.ch/R-manual/R-devel/library/stats/html/hclust.html (accessed on 20 July 2022).
- Centrality-Wikipedia. Available online: https://en.wikipedia.org/wiki/Centrality (accessed on 20 July 2022).
- Jeong, H.; Mason, S.P.; Barabási, A.-L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.X.; Wang, H.; Pan, Y. Identification of Essential Proteins from Weighted Protein-protein Interaction Networks. J. Bioinform. Comput. Biol. 2013, 11, 1341002. [Google Scholar] [CrossRef]
- Sabidussi, G. The Centrality Index of a Graph. Psychometrika 1966, 31, 581–603. [Google Scholar] [CrossRef] [PubMed]
- Anthonisse, J.M. The Rush in a Directed Graph; Stichting Mathematisch Centrum: Amsterdam, The Netherlands, 1971. [Google Scholar]
- Bonacich, P. Power and Centrality: A Family of Measures. Am. J. Sociol. 1987, 92, 1170–1182. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA Package FAQ. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/faq.html (accessed on 19 July 2022).
- Zheng, T.X.; Li, H.S.; Han, N.; Wang, S.Y.; Price, J.H.; Wang, M.Z.; Zhang, D.Y. Functional Characterization of Two Elongases of Very Long-Chain Fatty Acid from Tenebrio molitor L. (Coleoptera: Tenebrionidae). Sci. Rep. 2017, 7, 10990. [Google Scholar] [CrossRef] [PubMed]
- Li, D.T.; Chen, X.; Wang, X.Q.; Moussian, B.; Zhang, C.X. The Fatty Acid Elongase Gene Family in the Brown Planthopper, Nilaparvata lugens. Insect Biochem. Mol. Biol. 2019, 108, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Sassa, T.; Kihara, A. Metabolism of Very Long-Chain Fatty Acids: Genes and Pathophysiology. Biomol. Ther. 2014, 22, 83–92. [Google Scholar] [CrossRef]
- Iida, K.; Cavener, D.R. Glucose Dehydrogenase is Required for Normal Sperm Storage and Utilization in Female Drosophila melanogaster. J. Exp. Biol. 2004, 207, 675–681. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, W.-F.; Ling, X.-F.; Lu, Q.; Wang, W.-W.; Zhang, X.; Feng, Y.; Chen, X.-M.; Chen, H. Identification of the Key Pathways and Genes Involved in the Wax Biosynthesis of the Chinese White Wax Scale Insect (Ericerus pela Chavannes) by Integrated Weighted Gene Coexpression Network Analysis. Genes 2022, 13, 1364. https://doi.org/10.3390/genes13081364
Ding W-F, Ling X-F, Lu Q, Wang W-W, Zhang X, Feng Y, Chen X-M, Chen H. Identification of the Key Pathways and Genes Involved in the Wax Biosynthesis of the Chinese White Wax Scale Insect (Ericerus pela Chavannes) by Integrated Weighted Gene Coexpression Network Analysis. Genes. 2022; 13(8):1364. https://doi.org/10.3390/genes13081364
Chicago/Turabian StyleDing, Wei-Feng, Xiao-Fei Ling, Qin Lu, Wei-Wei Wang, Xin Zhang, Ying Feng, Xiao-Ming Chen, and Hang Chen. 2022. "Identification of the Key Pathways and Genes Involved in the Wax Biosynthesis of the Chinese White Wax Scale Insect (Ericerus pela Chavannes) by Integrated Weighted Gene Coexpression Network Analysis" Genes 13, no. 8: 1364. https://doi.org/10.3390/genes13081364
APA StyleDing, W.-F., Ling, X.-F., Lu, Q., Wang, W.-W., Zhang, X., Feng, Y., Chen, X.-M., & Chen, H. (2022). Identification of the Key Pathways and Genes Involved in the Wax Biosynthesis of the Chinese White Wax Scale Insect (Ericerus pela Chavannes) by Integrated Weighted Gene Coexpression Network Analysis. Genes, 13(8), 1364. https://doi.org/10.3390/genes13081364