
A Comparison of Bioinformatics Pipelines for Enrichment Illumina Next Generation Sequencing Systems in Detecting SARS-CoV-2 Virus Strains

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral RNA Extractions and Library Preparation for Whole Genome Sequencing
2.2. Patients’ Whole Genome Sequencing Data
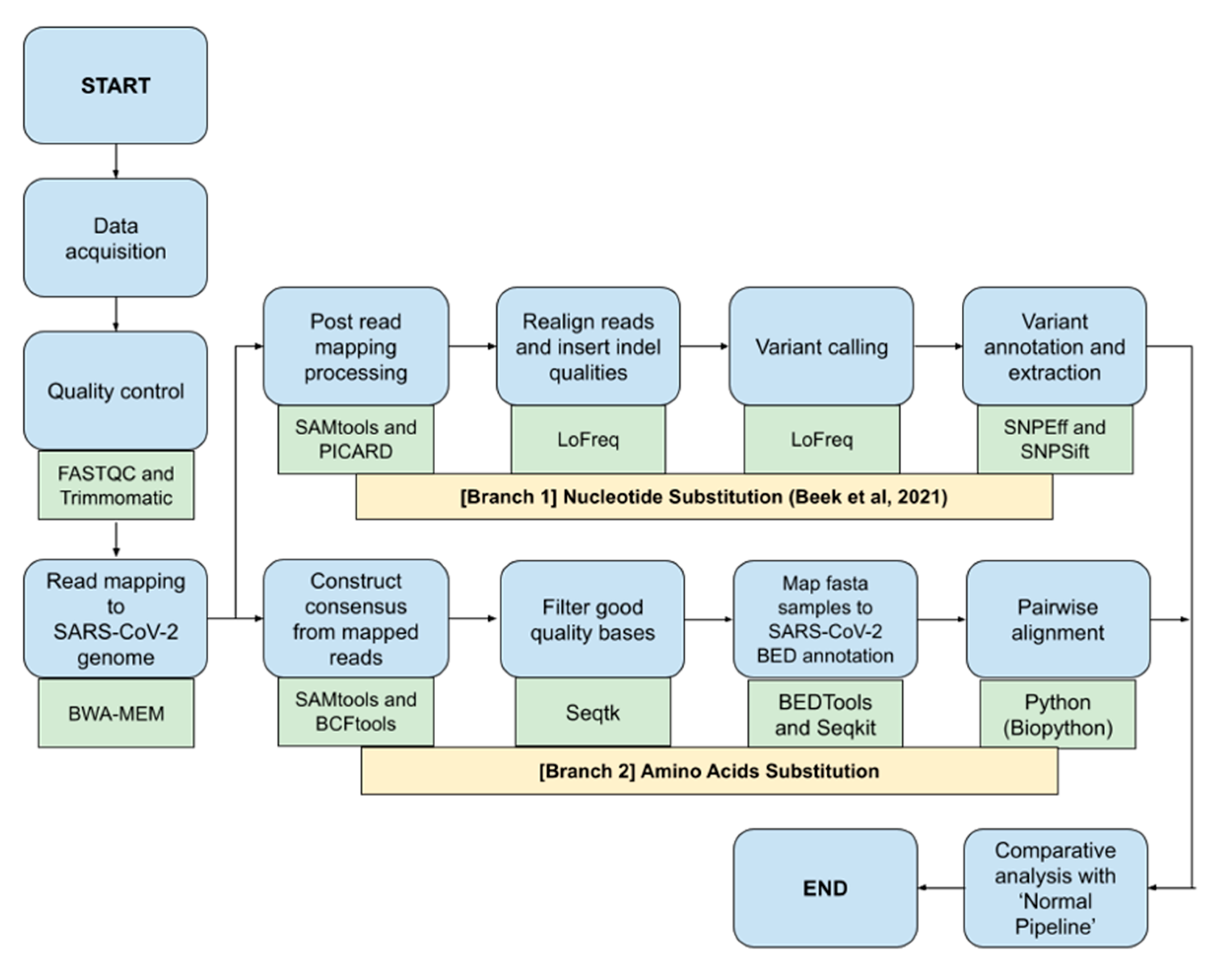
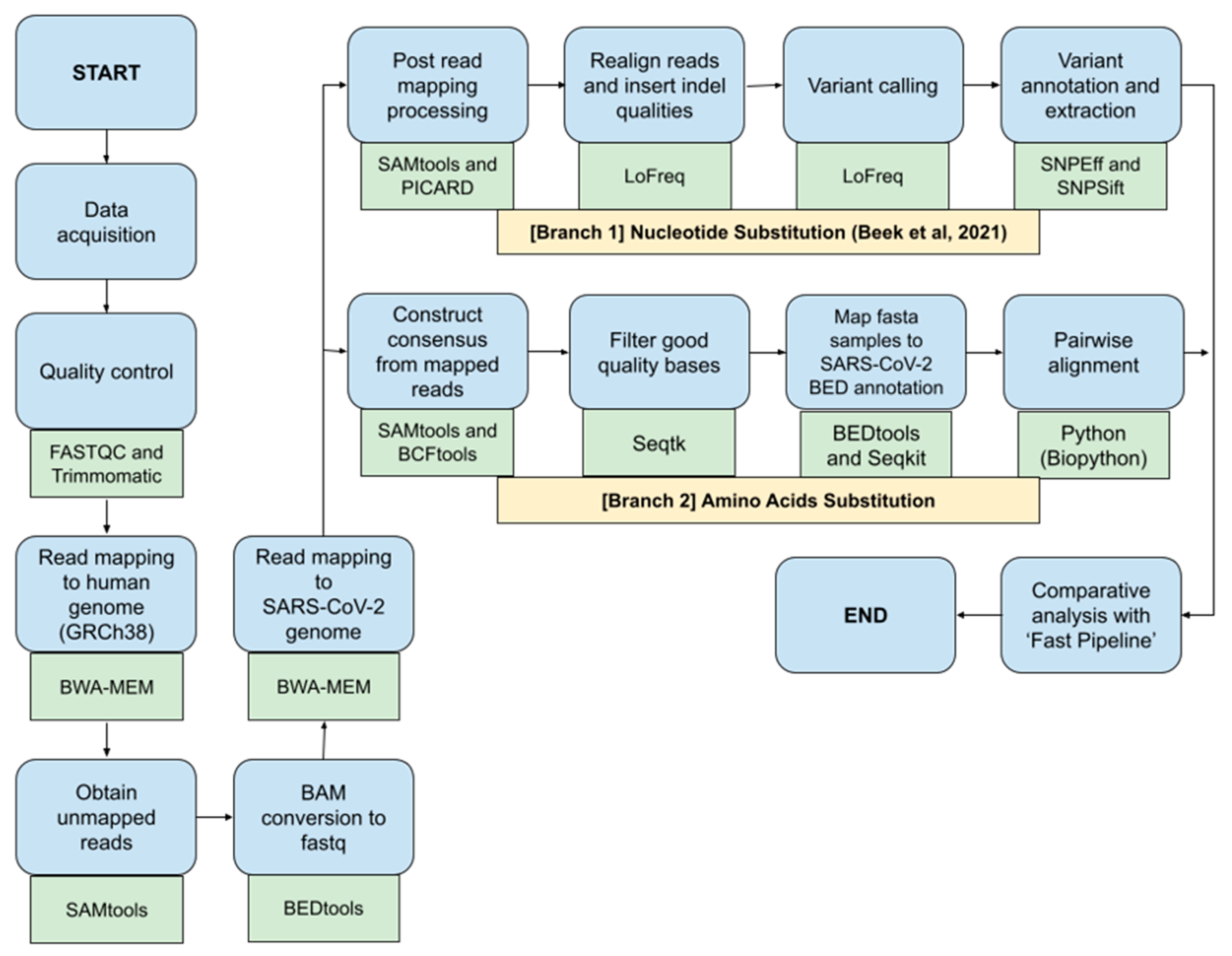
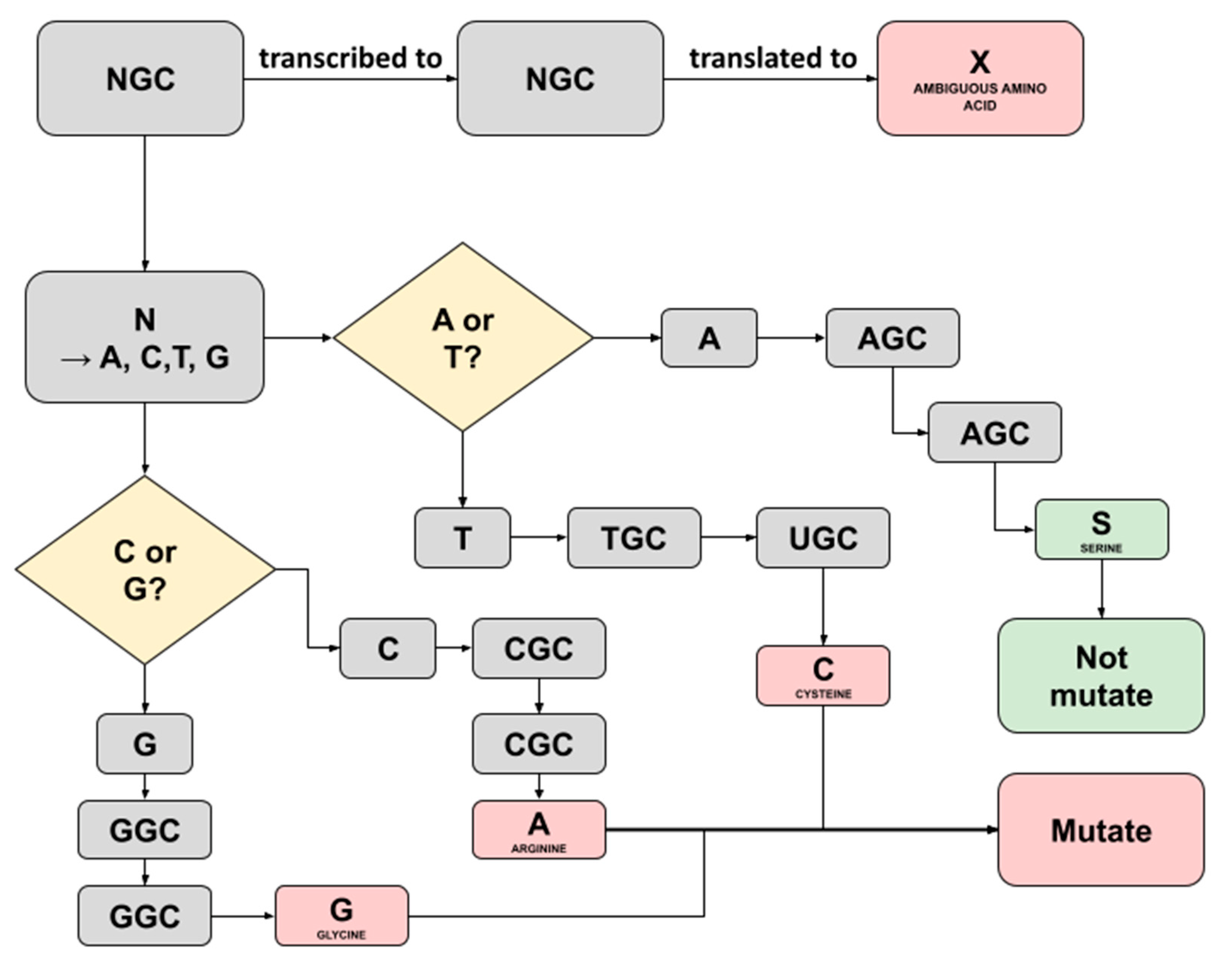
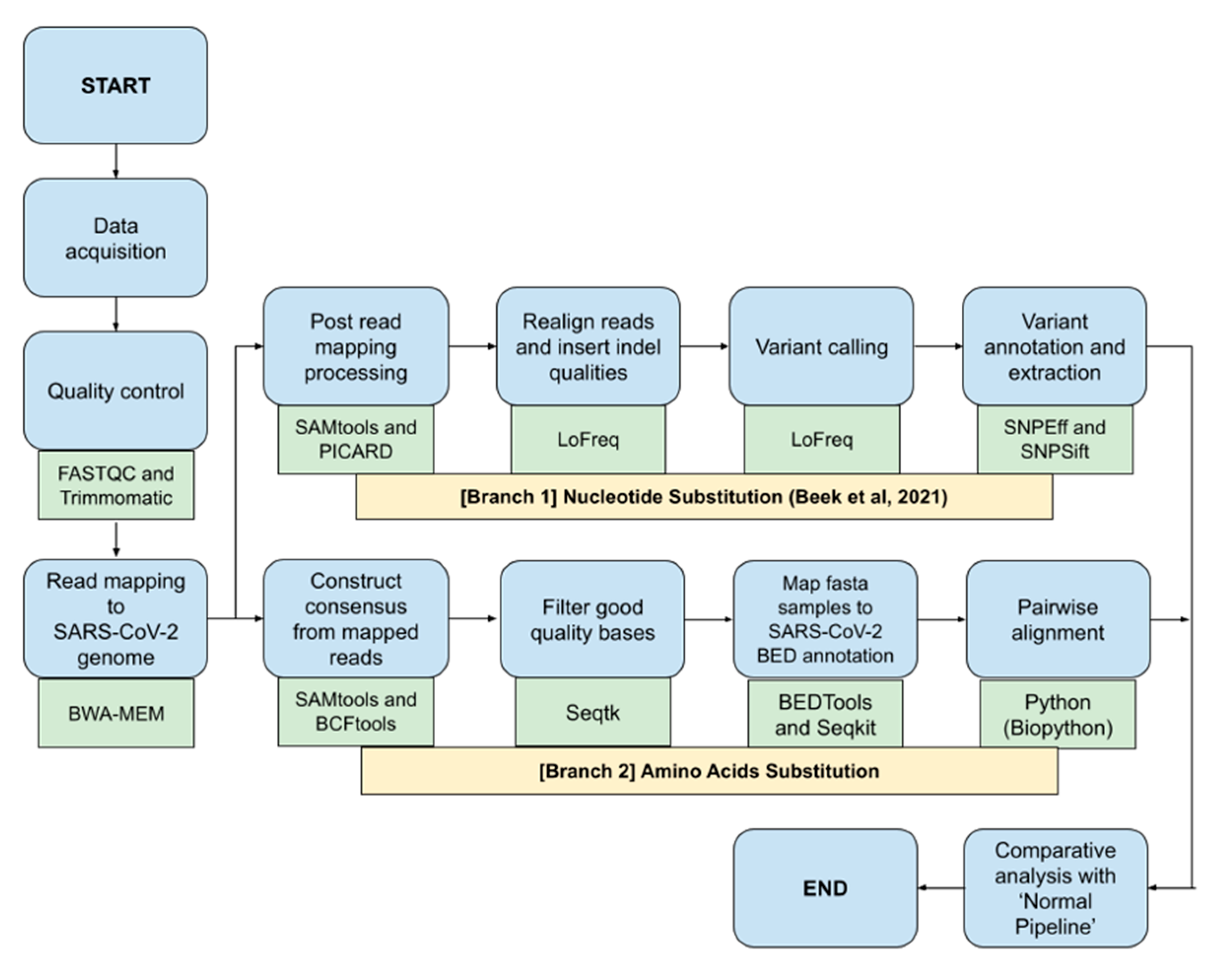
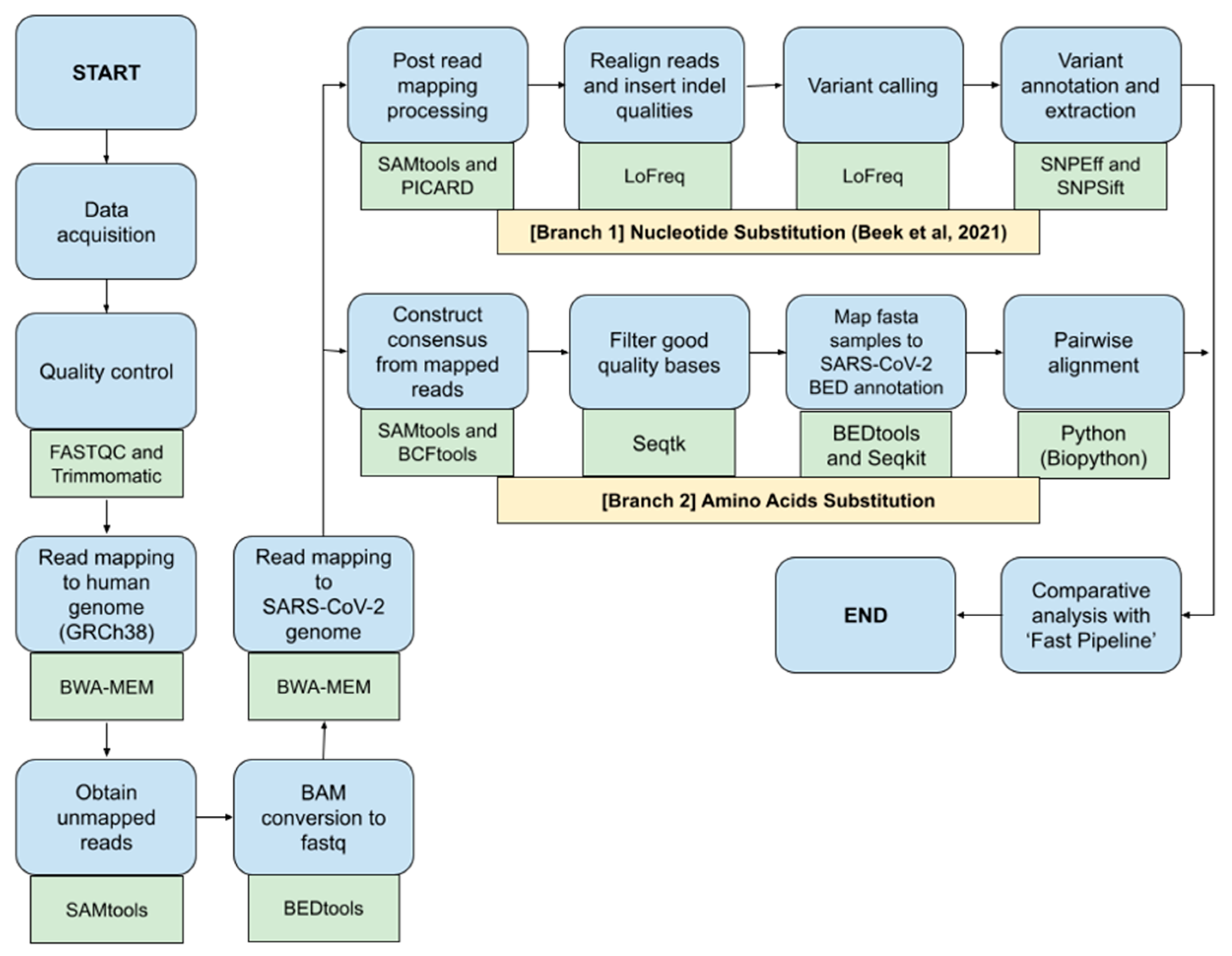
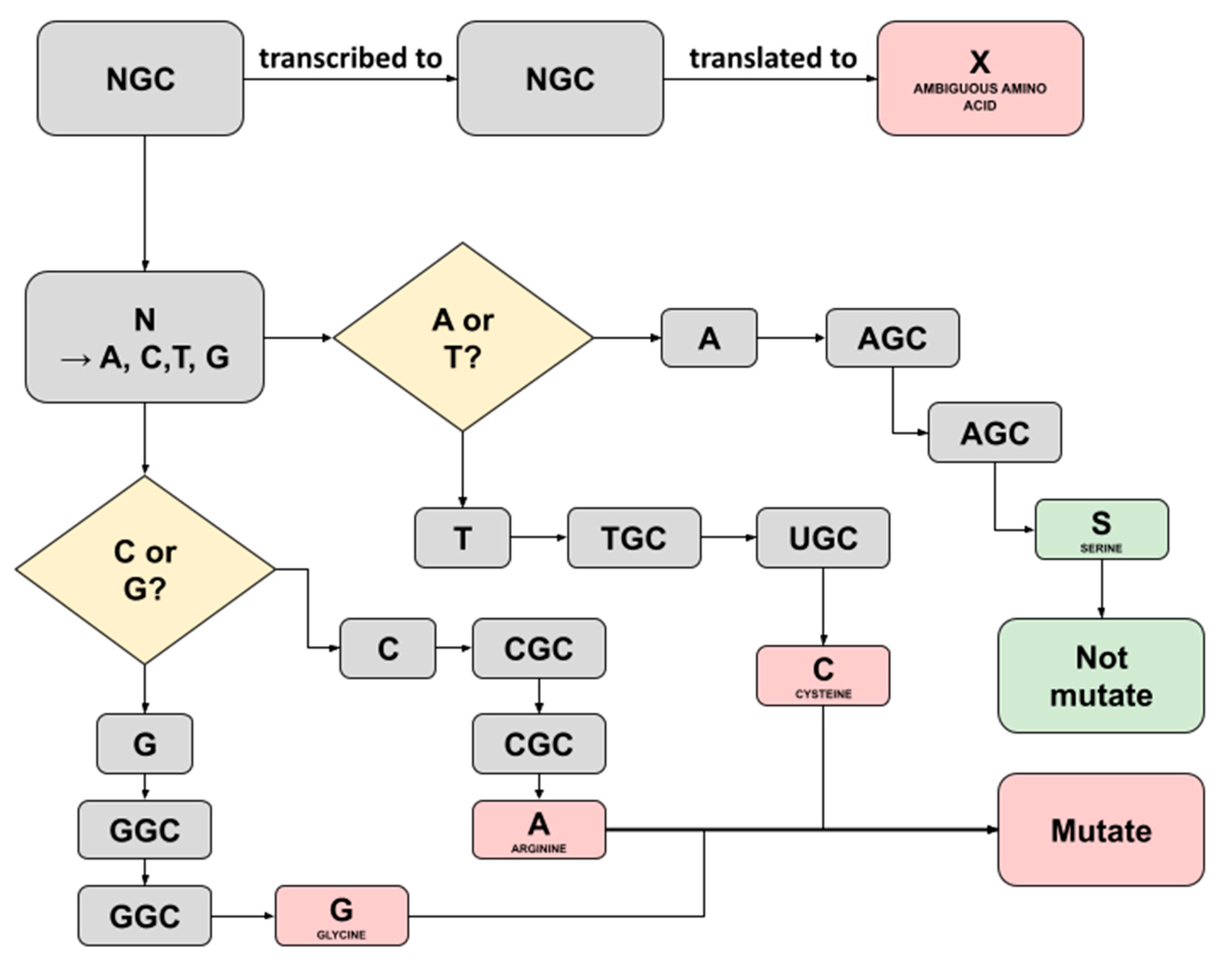
2.3. Bioinformatics Pipeline for SARS-CoV-2 Nucleotide and Amino Acids Variant Analysis
3. Results
3.1. Overview of Whole Genome Sequencing Data
3.2. Comparison of Reads Distribution in Normal Pipeline and Fast Pipeline
3.3. Comparison of Coverage Depth in Normal Pipeline and Fast Pipeline
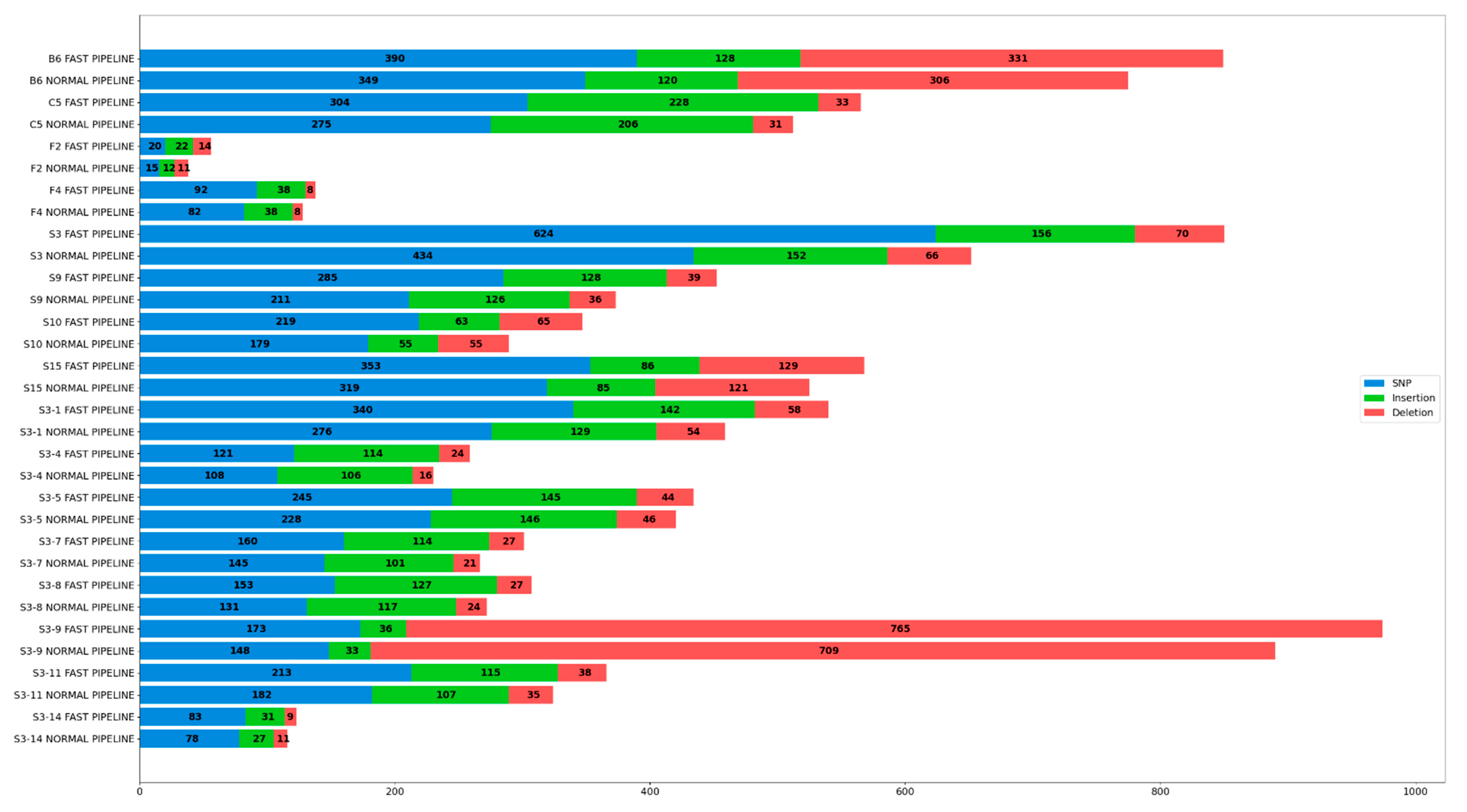
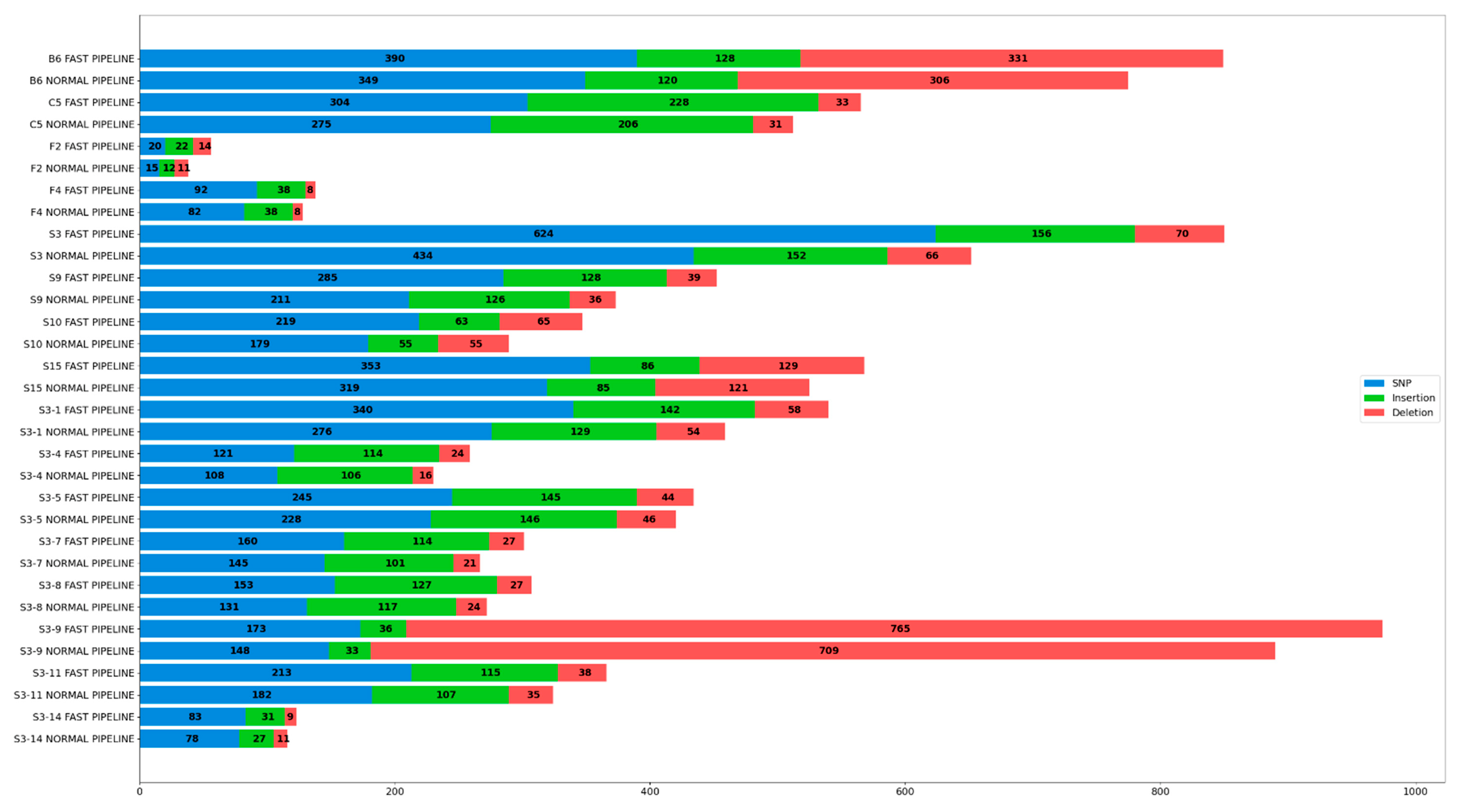
3.4. Comparison of Variations Annotated Post Variant Calling
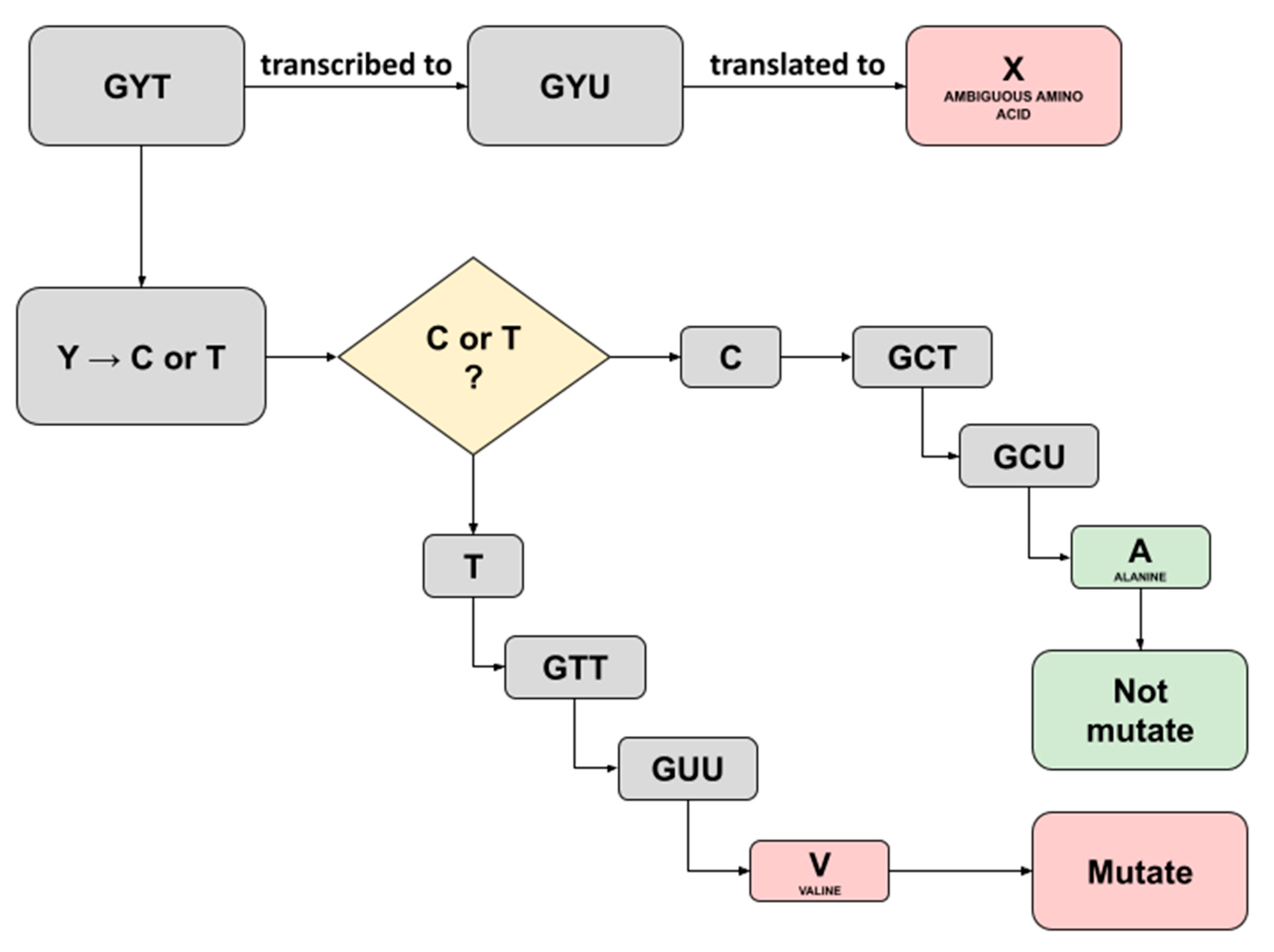
3.5. High Quality and Annotated Nucleotide Substitutions and Amino Acids Mutations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Nieuwenhuijse, D.F.; Stein, M.; O’Toole, Á.; Haverkate, M.; Mollers, M.; Kamga, S.K.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 2020, 26, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Enrichment Workflow for Detecting Coronavirus Using Illumina NGS Systems. 2020. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/appnotes/ngs-enrichment-coronavirus-app-note-1270-2020-002.pdf (accessed on 29 January 2021).
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed]
- Gunadi, H.W.; Marcellus, M.S.; Edwin Widyanto Daniwijaya, L.P.; Endah Supriyati, D.A.; Afiahayati, S.; Kristy Iskandar, N.A.; Alvin Santoso Kalim, D.A.; Kemala Athollah, E.A.; Titik Nuryastuti, T.W. Full-length genome characterization and phylogenetic analysis OF SARS-COV-2 virus strains from Yogyakarta and central Java, Indonesia. PeerJ 2020, 8, e10575. [Google Scholar] [CrossRef] [PubMed]
- Beek, M.; Clements, D.; Blankenberg, D.; Nekrutenko, A. Galaxy Training: From NCBI’s Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 Variant Analysis (Galaxy Training Materials). 2021. Available online: https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html (accessed on 1 March 2021).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Yang, S.-L.; Chen, G.-W.; Chen, Y.-W.; Huang, Y.-C.; Ning, H.-C.; Tsao, K.-C. A metagenomics study for the identification of respiratory viruses in mixed clinical specimens: An application of the iterative mapping approach. Arch. Virol. 2017, 162, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A method to identify respiratory virus infections in clinical samples using next-generation sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.B.; Thomson, E.C.; Hughes, J.; Aranday-Cortes, E.; McLauchlan, J.; da Silva Filipe, A.; Tong, L.; Manso, C.F.; Gifford, R.J.; Robertson, D.L.; et al. Interpreting Viral Deep Sequencing Data with GLUE. Viruses 2019, 11, 323. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.D. An extended IUPAC nomenclature code for polymorphic nucleic acids. Bioinformatics 2010, 26, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2019, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McAuley, A.J.; Kuiper, M.J.; Durr, P.A.; Bruce, M.P.; Barr, J.; Todd, S.; Au, G.G.; Blasdell, K.; Tachedjian, M.; Lowther, S.; et al. Experimental and in silico evidence suggests vaccines are unlikely to be affected by D614G mutation in SARS-CoV-2 spike protein. NPJ Vaccines 2020, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Miller, S.O.; Yen, C.H.; Wang, S.F. Impact of Genetic Variability in ACE2 Expression on the Evolutionary Dynamics of SARS-CoV-2 Spike D614G Mutation. Genes 2021, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Cahyani, I.; Putro, E.W.; Ridwanuloh, A.M.; Wibowo, S.; Hariyatun Syahputra, G.; Akbariani, G.; Utomo, A.R.; Ilyas, M.; Loose, M.; Kusharyoto, W. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements. Viruses 2022, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Hui, K.; Gu, H.; Ko, R.; Krishnan, P.; Ng, D.; Liu, G.Y.; Wan, C.K.; Cheung, M.C.; Ng, K.C.; et al. Introduction of ORF3a-Q57H SARS-CoV-2 Variant Causing Fourth Epidemic Wave of COVID-19, Hong Kong, China. Emerg. Infect. Dis. 2021, 27, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.G.; Hsiao, S.H.; Fann, Y.C.; Lee, Y.C. Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes 2022, 13, 686. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | NGS Sample Code | NGS Batch | Sample ID | Sex | Age (Years) | Collection Date | Comorbid |
|---|---|---|---|---|---|---|---|
| 1 | B6 | 1 | DIY-C25.2-02449 | Male | 77 | 22 June 2020 | Yes |
| 2 | C5 | 1 | DIY-C78.01481 | Female | 83 | 10 August 2020 | Yes |
| 3 | F2 | 1 | DIY-C25.2-00927 | Male | 30 | 16 May 2020 | No |
| 4 | F4 | 1 | KLN-C25.2-02538 | Female | 55 | 26 June 2020 | Yes |
| 5 | S3 | 2 | RSS-10001 | Male | 88 | 18 August 2020 | Yes |
| 6 | S9 | 2 | BBTKLPP-47964 | Male | 48 | 31 August 2020 | Yes |
| 7 | S10 | 2 | BBTKLPP-48651 | Male | 41 | 9 September 2020 | No |
| 8 | S15 | 2 | DIY-C78.00061 | Female | 49 | 16 June 2020 | No |
| 9 | S3-1 | 3 | DIY 1-58634 | Male | 65 | 18 September 2020 | Yes |
| 10 | S3-4 | 3 | DIY 1-24778 | Male | 34 | 23 December 2020 | No |
| 11 | S3-5 | 3 | DIY 1-10279 | Male | 77 | 7 September 2020 | No |
| 12 | S3-7 | 3 | DIY 1-10282 | Female | 42 | 7 September 2020 | No |
| 13 | S3-8 | 3 | DIY 1-24762 | Female | 48 | 23 December 2020 | No |
| 14 | S3-9 | 3 | RSS-10008 | Male | 58 | 27 December 2020 | Yes |
| 15 | S3-11 | 3 | DIY 1-24776 | Female | 34 | 23 December 2020 | No |
| 16 | S3-14 | 3 | 53311 | Female | 81 | 9 September 2020 | Yes |
| NGS Sample Code | Batch | CT Value | Total Sequences (Paired-End Reads) | Sequence Length (bp) | % GC |
|---|---|---|---|---|---|
| B6 | 1 | 19.70 | 11,268,022 | 35–74 | 41 |
| C5 | 1 | 16.90 | 2,707,228 | 35–74 | 42 |
| F2 | 1 | 27.92 | 2,461,478 | 35–74 | 50 |
| F4 | 1 | 24.68 | 1,366,538 | 35–74 | 45 |
| S3 | 2 | 18.10 | 18,807,934 | 35–74 | 38 |
| S9 | 2 | 19.64 | 7,827,098 | 35–74 | 46 |
| S10 | 2 | 21.24 | 2,698,396 | 35–74 | 42 |
| S15 | 2 | 22.31 | 6,111,408 | 35–74 | 46 |
| S3-1 | 3 | 19.53 | 3,566,896 | 35–74 | 40 |
| S3-4 | 3 | 13.27 | 1,167,562 | 35–74 | 38 |
| S3-5 | 3 | 21.00 | 9,941,746 | 35–74 | 38 |
| S3-7 | 3 | 21.55 | 1,669,316 | 35–74 | 39 |
| S3-8 | 3 | 15.67 | 2,731,486 | 35–74 | 39 |
| S3-9 | 3 | 22.27 | 4,748,810 | 35–74 | 45 |
| S3-11 | 3 | 16.89 | 5,895,626 | 35–74 | 39 |
| S3-14 | 3 | 17.73 | 376,514 | 35–74 | 44 |
| NGS Sample Code | Total Sequences | ||
|---|---|---|---|
| Before Trimming (Paired-End Reads) | Post-Trimming (QC) (Paired-End Reads) | Trimmed Sequence (%) | |
| B6 | 11,268,022 | 11,184,784 | 0.74 |
| C5 | 2,707,228 | 2,683,232 | 0.89 |
| F2 | 2,461,478 | 2,440,518 | 0.85 |
| F4 | 1,366,538 | 1,345,416 | 1.55 |
| S3 | 18,807,934 | 18,387,180 | 2.24 |
| S9 | 7,827,098 | 7,587,506 | 3.06 |
| S10 | 2,698,396 | 2,590,256 | 4.01 |
| S15 | 6,111,408 | 5,942,890 | 2.76 |
| S3-1 | 3,566,896 | 3,502,824 | 1.80 |
| S3-4 | 1,167,562 | 1,155,934 | 1.00 |
| S3-5 | 9,941,746 | 9,807,834 | 1.35 |
| S3-7 | 1,640,458 | 1,640,458 | 1.73 |
| S3-8 | 2,731,486 | 2,696,200 | 1.29 |
| S3-9 | 4,670,496 | 4,670,496 | 1.65 |
| S3-11 | 5,816,070 | 5,816,070 | 1.35 |
| S3-14 | 372,662 | 372,662 | 1.02 |
| Average | 1.70 | ||
| NGS Sample Code | Unmapped to Sars-Cov-2 Genome | Fully Mapped to Sars-Cov-2 Genome | ||
|---|---|---|---|---|
| Number of Reads | Percentage (%) | Number of Reads | Percentage (%) | |
| B6 | 2,028,393 | 18.14 | 9,156,391 | 81.86 |
| C5 | 1,108,537 | 41.31 | 1,574,695 | 58.69 |
| F2 | 2,391,210 | 97.98 | 49,308 | 2.02 |
| F4 | 1,203,004 | 89.42 | 142,412 | 10.58 |
| S3 | 548,965 | 2.99 | 17,838,215 | 97.01 |
| S9 | 4,969,736 | 65.50 | 2,617,770 | 34.50 |
| S10 | 1,452,990 | 56.09 | 1,137,266 | 43.91 |
| S15 | 4,071,831 | 68.52 | 1,871,059 | 31.48 |
| S3-1 | 830,959 | 23.72 | 2,671,865 | 76.28 |
| S3-4 | 19,722 | 1.71 | 1,136,212 | 98.29 |
| S3-5 | 205,582 | 2.10 | 9,602,252 | 97.90 |
| S3-7 | 275,337 | 16.78 | 1,365,121 | 83.22 |
| S3-8 | 175,137 | 6.50 | 2,521,063 | 93.50 |
| S3-9 | 3,427,102 | 73.38 | 1,243,394 | 26.62 |
| S3-11 | 576,452 | 9.91 | 5,239,618 | 90.09 |
| S3-14 | 290,681 | 78.00 | 81,981 | 22.00 |
| NGS Sample Code | Fully Mapped to Sars-Cov-2 Genome | Fully Mapped to Human Genome | Neither Both | Skipped during BAM to FASTQ Conversion | ||||
|---|---|---|---|---|---|---|---|---|
| Number of Reads | Percentage (%) | Number of Reads | Percentage (%) | Number of Reads | Percentage (%) | Number of Reads | Percentage (%) | |
| B6 | 8,743,980 | 78.18 | 2,435,133 | 21.77 | 3444 | 0.02 | 2227 | 0.02 |
| C5 | 1,467,402 | 54.69 | 1,125,429 | 41.94 | 89,534 | 3.34 | 867 | 0.03 |
| F2 | 38,668 | 1.58 | 2,399,180 | 98.31 | 2272 | 0.09 | 398 | 0.02 |
| F4 | 134,956 | 10.03 | 1,210,132 | 89.94 | 108 | 0.01 | 220 | 0.02 |
| S3 | 15,158,756 | 82.44 | 3,216,116 | 17.49 | 9700 | 0.05 | 2608 | 0.01 |
| S9 | 2,363,094 | 31.14 | 5,214,314 | 68.72 | 7000 | 0.09 | 3098 | 0.04 |
| S10 | 1,009,265 | 38.96 | 1,546,174 | 59.69 | 34,167 | 1.32 | 650 | 0.03 |
| S15 | 1,676,134 | 28.20 | 4,235,721 | 71.27 | 28,482 | 0.48 | 2553 | 0.04 |
| S3-1 | 2,321,562 | 66.28 | 1,180,022 | 33.69 | 502 | 0.01 | 738 | 0.02 |
| S3-4 | 988,416 | 85.51 | 165,770 | 14.34 | 1706 | 0.15 | 42 | 0.00 |
| S3-5 | 8,996,852 | 91.73 | 807,254 | 8.23 | 3020 | 0.03 | 708 | 0.01 |
| S3-7 | 1,249,452 | 76.16 | 389,999 | 23.77 | 512 | 0.03 | 495 | 0.03 |
| S3-8 | 2,332,361 | 86.51 | 345,223 | 12.80 | 18,489 | 0.69 | 127 | 0.00 |
| S3-9 | 1,156,467 | 24.76 | 3,230,410 | 69.17 | 282,417 | 6.05 | 1202 | 0.03 |
| S3-11 | 4,628,769 | 79.59 | 1,178,473 | 20.26 | 8509 | 0.15 | 319 | 0.01 |
| S3-14 | 76,805 | 20.61 | 295,584 | 79.32 | 207 | 0.06 | 66 | 0.02 |
| Average | 53.52 | Average | 45.67 | Average | 0.78 | Average | 0.02 | |
| NGS Sample Code | Read Mapping Coverage (Times) | Difference Fast vs. Normal (%) | |
|---|---|---|---|
| Fast Pipeline | Normal Pipeline | ||
| B6 | 22,352.5 | 21,357.2 | 4.70 |
| C5 | 3833.9 | 3576.5 | 7.20 |
| F2 | 115 | 94.6 | 21.6 |
| F4 | 347.7 | 329.8 | 5.40 |
| S3 | 43,244.4 | 36,843 | 17.4 |
| S9 | 6350.02 | 5744.18 | 10.5 |
| S10 | 2756.31 | 2457.99 | 12.1 |
| S15 | 4545.72 | 4077.32 | 11.5 |
| S3-1 | 6494.44 | 5653.07 | 14.9 |
| S3-4 | 2764.01 | 2410.1 | 14.7 |
| S3-5 | 23,481.7 | 22,016.8 | 6.60 |
| S3-7 | 3333.82 | 3054.74 | 9.10 |
| S3-8 | 6163.56 | 5706.29 | 8.00 |
| S3-9 | 3033.52 | 2824.06 | 7.40 |
| S3-11 | 12,794.7 | 11,323.3 | 13.00 |
| S3-14 | 199.371 | 186.96 | 6.60 |
| Average | 10.66 | ||
| NGS Sample Code | Fast Pipeline | Normal Pipeline | Difference Fast vs. Normal (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #SNP | #Insertion | #Deletion | All Variation | #SNP | #Insertion | #Deletion | All Variation | #SNP | #Insertion | #Deletion | All Variation | |
| B6 | 390 | 128 | 331 | 849 | 349 | 120 | 306 | 775 | 11.75 | 6.67 | 8.17 | 9.55 |
| C5 | 304 | 228 | 33 | 565 | 275 | 206 | 31 | 512 | 10.55 | 10.68 | 6.45 | 10.35 |
| F2 | 20 | 22 | 14 | 56 | 15 | 12 | 11 | 38 | 33.33 | 83.33 | 27.27 | 47.37 |
| F4 | 92 | 38 | 8 | 138 | 82 | 38 | 8 | 128 | 12.20 | 0.00 | 0.00 | 7.81 |
| S3 | 624 | 156 | 70 | 850 | 434 | 152 | 66 | 652 | 43.78 | 2.63 | 6.06 | 30.37 |
| S9 | 285 | 128 | 39 | 452 | 211 | 126 | 36 | 373 | 35.07 | 1.59 | 8.33 | 21.18 |
| S10 | 219 | 63 | 65 | 347 | 179 | 55 | 55 | 289 | 22.35 | 14.55 | 18.18 | 20.07 |
| S15 | 353 | 86 | 129 | 568 | 319 | 85 | 121 | 525 | 10.66 | 1.18 | 6.61 | 8.19 |
| S3-1 | 340 | 142 | 58 | 540 | 276 | 129 | 54 | 459 | 23.19 | 10.08 | 7.41 | 17.65 |
| S3-4 | 121 | 114 | 24 | 259 | 108 | 106 | 16 | 230 | 12.04 | 7.55 | 50.00 | 12.61 |
| S3-5 | 245 | 145 | 44 | 434 | 228 | 146 | 46 | 420 | 7.46 | −0.68 | −4.35 | 3.33 |
| S3-7 | 160 | 114 | 27 | 301 | 145 | 101 | 21 | 267 | 10.34 | 12.87 | 28.57 | 12.73 |
| S3-8 | 153 | 127 | 27 | 307 | 131 | 117 | 24 | 272 | 16.79 | 8.55 | 12.50 | 12.87 |
| S3-9 | 173 | 36 | 765 | 974 | 148 | 33 | 709 | 890 | 16.89 | 9.09 | 7.90 | 9.44 |
| S3-11 | 213 | 115 | 38 | 366 | 182 | 107 | 35 | 324 | 17.03 | 7.48 | 8.57 | 12.96 |
| S3-14 | 83 | 31 | 9 | 123 | 78 | 27 | 11 | 116 | 6.41 | 14.81 | −18.18 | 6.03 |
| Average | 18.11 | 11.90 | 10.84 | 15.16 | ||||||||
| POSITION | 5’UTR | NSP3-ORF1AB | NSP5-ORF1AB | NSP12-ORF1AB | NSP13-ORF1AB | NSP14-ORF1AB | SPIKE-S | NS3-ORF3A | MATRIX-M | NS7A-ORF7A | NP-N | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POSITION | 241 | 3037 | 3529 | 4754 | 5184 | 10201 | 10507 | 14055 | 14292 | 14408 | 14694 | 15406 | 17964 | 18744 | 18877 | 23403 | 25553 | 25563 | 25687 | 26735 | 26867 | 27610 | 28735 | 28752 | 29209 |
| REFERENCE (NC_045512.2) | C | C | T | C | C | G | C | G | C | C | C | G | G | C | C | A | C | G | G | C | A | C | T | A | A |
| B6 FAST PIPELINE | T | T | T | C | T | G | T | G | C | T | C | G | G | T | T | G | C | T | G | T | G | C | T | A | A |
| B6 NORMAL PIPELINE | T | T | T | C | T | G | T | G | C | T | C | G | G | T | T | G | C | T | G | T | G | C | T | A | A |
| C5 FAST PIPELINE | T | T | C | T | C | G | C | G | T | T | T | T | T | C | T | G | T | T | T | T | A | C | C | G | A |
| C5 NORMAL PIPELINE | T | T | C | T | C | G | C | G | T | T | T | T | T | C | T | G | T | T | T | T | A | C | C | G | A |
| F2 FAST PIPELINE | C | C | T | C | C | T | C | G | C | C | C | G | G | C | C | A | C | G | G | C | A | C | T | A | G |
| F2 NORMAL PIPELINE | C | C | T | C | C | T | C | G | C | C | C | G | G | C | C | A | C | G | G | C | A | C | T | A | G |
| F4 FAST PIPELINE | T | T | T | C | T | G | T | T | C | T | C | G | G | T | T | G | C | T | G | T | G | T | T | A | A |
| F4 NORMAL PIPELINE | T | T | T | C | T | G | T | T | C | T | C | G | G | T | T | G | C | T | G | T | G | T | T | A | A |
| (a) Identified SNPs in All Batch 2 Samples Running All Pipelines (Part 1) | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REGION | 5’UTR | NSP1-ORF1AB | NSP3-ORF1AB | NSP5-ORF1AB | NSP6-ORF1AB | NSP8-ORF1AB | NSP9-ORF1AB | NSP12-ORF1AB | NSP13-ORF1AB | |||||||||||||||||
| POSITION | 241 | 1545 | 2263 | 2512 | 3037 | 4084 | 5184 | 5784 | 6312 | 7639 | 10089 | 10507 | 11083 | 12152 | 12439 | 12809 | 13730 | 14120 | 14183 | 14408 | 15543 | 15765 | 16156 | 16395 | 16647 | 16694 |
| REFERENCE (NC_045512.2) | C | C | C | A | C | C | C | C | C | C | A | C | G | G | C | C | C | C | C | C | G | A | A | A | G | C |
| S3 FAST PIPELINE | T | T | C | A | T | T | T | C | C | C | G | T | G | G | C | C | C | C | T | T | G | A | A | T | T | C |
| S3 NORMAL PIPELINE | T | T | C | A | T | T | T | C | C | C | G | T | G | G | C | C | C | C | T | T | G | A | A | T | T | C |
| S9 FAST PIPELINE | C | C | C | G | C | C | C | C | A | C | A | C | T | A | T | T | T | C | C | C | G | A | G | A | G | T |
| S9 NORMAL PIPELINE | C | C | C | G | C | C | C | C | A | C | A | C | T | A | T | T | T | C | C | C | G | A | G | A | G | T |
| S10 FAST PIPELINE | T | C | C | A | T | C | C | C | C | C | A | C | G | G | C | C | C | T | C | T | G | G | A | A | G | C |
| S10 NORMAL PIPELINE | T | C | C | A | T | C | C | C | C | C | A | C | G | G | C | C | C | T | C | T | G | G | A | A | G | C |
| S15 FAST PIPELINE | T | C | T | A | T | C | T | T | C | T | A | T | G | G | C | C | C | C | C | T | T | A | A | A | G | C |
| S15 NORMAL PIPELINE | T | C | T | A | T | C | T | T | C | T | A | T | G | G | C | C | C | C | C | T | T | A | A | A | G | C |
| (b) Identified SNPs in All Batch 2 Samples Running All Pipelines (Part 2) | ||||||||||||||||||||||||||
| REGION | NSP14-ORF1AB | NSP15-A1-ORF1AB | SPIKE-S | NS3-ORF3A | MATRIX-M | ORF8 | NP-N | ORF10 | ||||||||||||||||||
| POSITION | 18744 | 18877 | 19002 | 20124 | 21652 | 21742 | 21748 | 21809 | 22200 | 22334 | 23403 | 23593 | 23929 | 25563 | 26056 | 26735 | 26867 | 28073 | 28311 | 28628 | 28851 | 28975 | 29642 | |||
| REFERENCE (NC_045512.2) | C | C | A | T | T | C | T | G | T | T | A | G | C | G | G | C | A | G | C | G | G | G | C | |||
| S3 FAST PIPELINE | T | T | A | T | T | T | T | G | C | T | G | G | C | T | G | T | G | G | C | T | T | G | C | |||
| S3 NORMAL PIPELINE | T | T | A | T | T | T | T | G | C | T | G | G | C | T | G | T | G | G | C | T | T | G | C | |||
| S9 FAST PIPELINE | C | C | G | C | C | C | C | G | T | C | G | G | T | G | G | C | A | A | T | G | G | G | C | |||
| S9 NORMAL PIPELINE | C | C | G | C | C | C | C | G | T | C | G | G | T | G | G | C | A | A | T | G | G | G | C | |||
| S10 FAST PIPELINE | C | T | A | T | T | C | T | C | T | T | G | T | C | T | T | T | A | G | C | G | G | T | T | |||
| S10 NORMAL PIPELINE | C | T | A | T | T | C | T | C | T | T | G | T | C | T | T | T | A | G | C | G | G | T | T | |||
| S15 FAST PIPELINE | T | T | A | T | T | C | T | G | T | T | G | G | C | T | G | T | A | G | C | G | G | G | C | |||
| S15 NORMAL PIPELINE | T | T | A | T | T | C | T | G | T | T | G | G | C | T | G | T | A | G | C | G | G | G | C | |||
| REGION | NSP3-ORF1AB | NSP4-ORF1AB | NSP5A-ORF1AB | NSP6-ORF1AB | NSP7-ORF1AB | NSP12-ORF1AB | NSP15-ORF1AB | SPIKE GLYCOPROTEIN | ORF3A | ORF8 | NP-N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POSITION | 3305 | 5184 | 5554 | 6309 | 6906 | 9701 | 9710 | 9711 | 10313 | 10904 | 10995 | 11219 | 11991 | 14120 | 14408 | 14741 | 15848 | 19793 | 19794 | 20443 | 20611 | 21575 | 22200 | 23042 | 23270 | 23403 | 23599 | 23629 | 25337 | 25563 | 25590 | 25904 | 28020 | 28628 | 28655 | 28724 | 28851 | 28881 | 28883 | 28975 | 28977 |
| REFERENCE (NC_045512.2) | A | C | G | G | C | A | T | C | C | A | A | A | A | C | C | C | C | G | G | G | C | C | T | T | G | A | T | T | G | G | A | C | T | G | G | C | G | G | G | G | C |
| S3-1 FAST PIPELINE | A | T | G | G | C | A | T | C | C | A | A | A | A | C | T | T | C | G | G | T | T | C | C | T | G | G | T | T | G | T | A | C | T | T | G | C | T | G | G | G | C |
| S3-1 NORMAL PIPELINE | A | T | G | G | C | A | T | C | C | A | A | A | A | C | T | T | C | G | G | T | T | C | C | T | G | G | T | T | G | T | A | C | T | T | G | C | T | G | G | G | C |
| S3-4 FAST PIPELINE | A | C | G | G | C | A | A | A | T | A | A | A | A | T | T | C | T | G | G | G | C | C | T | T | T | G | A | T | T | T | A | C | T | G | G | C | G | G | G | G | T |
| S3-4 NORMAL PIPELINE | A | C | G | G | C | A | A | A | T | A | A | A | A | T | T | C | T | G | G | G | C | C | T | T | T | G | A | T | T | T | A | C | T | G | G | C | G | G | G | G | T |
| S3-5 FAST PIPELINE | C | C | G | G | C | G | T | C | C | A | A | A | A | T | T | C | T | G | G | G | C | T | T | T | G | G | T | T | G | T | T | T | T | G | T | C | G | G | G | G | T |
| S3-5 NORMAL PIPELINE | C | C | G | G | C | G | T | C | C | A | A | A | A | T | T | C | T | G | G | G | C | T | T | T | G | G | T | T | G | T | T | T | T | G | T | C | G | G | G | G | T |
| S3-7 FAST PIPELINE | C | C | G | G | C | G | T | C | C | A | A | A | A | T | T | C | T | G | G | G | C | T | T | T | G | G | T | T | G | T | T | T | T | G | T | C | G | G | G | G | T |
| S3-7 NORMAL PIPELINE | C | C | G | G | C | G | T | C | C | A | A | A | A | T | T | C | T | G | G | G | C | T | T | T | G | G | T | T | G | T | T | T | T | G | T | C | G | G | G | G | T |
| S3-8 FAST PIPELINE | A | T | G | C | C | A | T | C | C | A | A | G | A | C | T | C | C | G | G | G | C | C | T | C | G | G | T | G | G | T | A | C | T | T | G | C | G | G | G | T | C |
| S3-8 NORMAL PIPELINE | A | T | G | C | C | A | T | C | C | A | A | G | A | C | T | C | C | G | G | G | C | C | T | C | G | G | T | G | G | T | A | C | T | T | G | C | G | G | G | T | C |
| S3-9 FAST PIPELINE | A | C | T | G | T | A | T | C | C | G | G | A | G | C | T | C | C | T | T | G | C | C | T | T | G | G | T | T | G | G | A | C | T | G | G | C | G | A | C | G | C |
| S3-9 NORMAL PIPELINE | A | C | T | G | C | A | T | C | C | G | G | A | G | C | T | C | C | T | T | G | C | C | T | T | G | G | T | T | G | G | A | C | C | G | G | T | G | A | C | G | C |
| S3-11 FAST PIPELINE | A | C | G | G | C | A | A | A | T | A | A | A | A | T | T | C | T | G | G | G | C | C | T | T | T | G | A | T | T | T | A | C | T | G | G | C | G | G | G | G | T |
| S3-11 NORMAL PIPELINE | A | C | G | G | C | A | A | A | T | A | A | A | A | T | T | C | T | G | G | G | C | C | T | T | T | G | A | T | T | T | A | C | T | G | G | C | G | G | G | G | T |
| S3-14 FAST PIPELINE | A | T | G | G | C | A | T | C | C | A | A | A | A | C | C | C | C | G | G | G | C | T | T | T | G | G | T | T | G | T | A | C | T | T | G | C | T | G | G | G | C |
| S3-14 NORMAL PIPELINE | A | T | G | G | C | A | T | C | C | A | A | A | A | C | C | C | C | G | G | G | C | T | T | T | G | G | T | T | G | G | A | C | T | T | G | C | T | G | G | G | C |
| NGS Sample Code | Length of Consensus Sequence (bp) | |
|---|---|---|
| Fast Pipeline | Normal Pipeline | |
| B6 | 29,903 | 29,894 |
| C5 | 29,903 | 29,892 |
| F2 | 29,903 | 29,853 |
| F4 | 29,903 | 29,877 |
| S3 | 29,903 | 29,890 |
| S9 | 29,903 | 29,892 |
| S10 | 29,903 | 29,870 |
| S15 | 29,903 | 29,879 |
| S3-1 | 29,903 | 29,892 |
| S3-4 | 29,903 | 29,870 |
| S3-5 | 29,903 | 29,877 |
| S3-7 | 29,903 | 29,867 |
| S3-8 | 29,903 | 29,870 |
| S3-9 | 29,903 | 29,892 |
| S3-11 | 29,903 | 29,870 |
| S3-14 | 29,903 | 29,869 |
| REGION | 5’UTR | NSP3-ORF1AB | NSP5-ORF1AB | NSP12-ORF1AB | NSP13-ORF1AB | SPIKE-S | NS3-ORF3A | NS7A-ORF7A | NP-N | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POSITION | 81 | 679 | 822 | 49 | 314 | 646 | 769 | 576 | 614 | 54 | 57 | 99 | 73 | 160 |
| REFERENCE (NC_045512.2) | R | P | P | M | P | A | S | M | D | A | Q | A | H | Q |
| B6 FAST PIPELINE | C | P | L | M | L | A | S | M | G | A | H | A | H | Q |
| B6 NORMAL PIPELINE | C | P | L | M | L | A | S | M | G | A | H | A | H | Q |
| C5 FAST PIPELINE | C | S | P | M | L | S | S | I | G | X | H | S | H | R |
| C5 NORMAL PIPELINE | C | S | P | M | L | S | S | I | G | X | H | S | H | R |
| F2 FAST PIPELINE | R | P | P | I | P | A | S | M | D | A | Q | A | H | Q |
| F2 NORMAL PIPELINE | R | P | P | I | P | A | X | M | D | A | Q | A | H | Q |
| F4 FAST PIPELINE | R | P | L | M | L | A | S | M | G | A | H | A | Y | Q |
| F4 NORMAL PIPELINE | R | P | L | M | L | A | S | M | G | A | H | A | Y | Q |
| REGION | 5’UTR | NSP3-ORF1AB | NSP5-ORF1AB | NSP6 | NSP8-ORF1AB | NSP9 | NSP12-ORF1AB | NSP13-ORF1AB | SPIKE-S | NS3-ORF3A | NP-N | ORF8 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POSITION | 81 | 822 | 1022 | 1198 | 12 | 37 | 21 | 42 | 88 | 218 | 239 | 314 | 897 | 153 | 83 | 213 | 258 | 614 | 677 | 57 | 222 | 13 | 119 | 193 | 234 | 29 |
| REFERENCE (NC_045512.2) | R | P | T | T | K | L | A | L | A | P | T | P | M | T | V | V | W | D | Q | Q | D | P | A | S | M | Q |
| S3 FAST PIPELINE | C | L | T | T | R | L | A | L | A | P | I | L | M | T | V | A | W | G | Q | H | D | P | S | I | M | Q |
| S3 NORMAL PIPELINE | C | L | T | T | R | L | A | L | A | P | I | L | M | T | V | A | W | G | Q | H | D | P | S | I | M | Q |
| S9 FAST PIPELINE | R | P | T | K | K | F | T | F | V | P | T | P | V | I | V | V | R | G | Q | Q | D | L | A | S | M | Q |
| S9 NORMAL PIPELINE | R | P | T | K | K | F | T | F | V | P | T | P | V | I | V | V | R | G | Q | Q | D | L | A | S | M | Q |
| S10 FAST PIPELINE | C | P | T | T | K | L | A | L | A | L | T | L | M | T | L | V | W | G | H | H | Y | P | A | S | I | * |
| S10 NORMAL PIPELINE | C | P | T | T | K | L | A | L | A | L | T | L | M | T | L | V | W | G | H | H | Y | P | A | S | I | * |
| S15 FAST PIPELINE | C | L | I | T | K | L | A | L | A | P | T | L | M | T | V | V | W | G | Q | H | D | P | A | S | M | Q |
| S15 NORMAL PIPELINE | C | L | I | T | K | L | A | L | A | P | T | L | M | T | V | V | W | G | Q | H | D | P | A | S | M | Q |
| REGION | NSP3-ORF1AB | NSP4-ORF1AB | NSP5A-ORF1AB | NSP6-ORF1AB | NSP7-ORF1AB | NSP12-ORF1AB | NSP15-ORF1AB | SPIKE GLYCOPROTEIN | ORF3A | ORF8 | NP-N | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POSITION | 196 | 822 | 945 | 1197 | 1369 | 383 | 386 | 87 | 284 | 8 | 83 | 50 | 227 | 323 | 434 | 803 | 58 | 275 | 331 | 5 | 213 | 494 | 570 | 614 | 679 | 689 | 1259 | 57 | 66 | 171 | 43 | 119 | 128 | 151 | 193 | 203 | 204 | 234 | 235 |
| REFERENCE (NC_045512.2) | M | P | K | S | S | I | S | L | S | K | M | E | P | P | S | T | W | V | L | L | V | S | A | D | N | S | D | Q | K | S | S | A | D | P | S | R | G | M | S |
| S3-1 FAST PIPELINE | M | L | K | S | S | I | S | L | S | K | M | E | P | L | F | T | W | F | F | L | A | S | A | G | N | S | D | H | K | S | S | S | D | P | I | R | G | M | S |
| S3-1 NORMAL PIPELINE | M | L | K | S | S | I | S | L | S | K | M | E | P | L | F | T | W | F | F | L | A | S | A | G | N | S | D | H | K | S | S | S | D | P | I | R | G | M | S |
| S3-4 FAST PIPELINE | M | P | K | S | S | I | T | F | S | K | M | E | L | L | S | I | W | V | L | L | V | S | S | G | K | S | Y | H | K | S | S | A | D | P | S | R | G | M | F |
| S3-4 NORMAL PIPELINE | M | P | K | S | S | I | T/Y | F | S | K | M | E | L | L | S | I | W | V | L | L | V | S | S | G | K | S | Y | H | K | S | S | A | D | P | S | R | G | M | F |
| S3-5 FAST PIPELINE | L | P | K | S | S | V | S | L | S | K | M | E | L | L | S | I | W | V | L | F | V | S | A | G | N | S | D | H | N | L | S | A | Y | P | S | R | G | M | F |
| S3-5 NORMAL PIPELINE | L | P | K | S | S | V | S | L | S | K | M | E | L | L | S | I | W | V | L | F | V | S | A | G | N | S | D | H | N | L | S | A | Y | P | S | R | G | M | F |
| S3-7 FAST PIPELINE | L | P | K | S | S | V | S | L | S | K | M | E | L | L | S | I | W | V | L | F | V | S | A | G | N | S | D | H | N | L | S | A | Y | P | S | R | G | M | F |
| S3-7 NORMAL PIPELINE | L | P | K | S | S | V | S | L | S | K | M | E | L | L | S | I | W | V | L | F | V | S | A | G | N | S | D | H | N | L | S | A | Y | P | S | R | G | M | F |
| S3-8 FAST PIPELINE | M | L | K | T | S | I | S | L | S | K | V | E | P | L | S | T | W | V | L | L | V | P | A | G | N | R | D | H | K | S | S | S | D | P | S | R | G | I | S |
| S3-8 NORMAL PIPELINE | M | L | K | T | S | I | S | L | S | K | V | E | P | L | S | T | W | V | L | L | V | P | A | G | N | R | D | H | K | S | S | S | D | P | S | R | G | I | S |
| S3-9 FAST PIPELINE | M | P | N | S | L | I | S | L | G | R | M | G | P | L | S | T | L/C | V | L | L | V | S | A | G | N | S | D | Q | K | S | S | A | D | P | S | K | R | M | S |
| S3-9 NORMAL PIPELINE | M | P | N | S | S | I | S | L | G | R | M | G | P | L | S | T | L/C | V | L | L | V | S | A | G | N | S | D | Q | K | S | P | A | D | S | S | K | R | M | S |
| S3-11 FAST PIPELINE | M | P | K | S | S | I | T/Y | F | S | K | M | E | L | L | S | I | W | V | L | L | V | S | S | G | K | S | Y | H | K | S | S | A | D | P | S | R | G | M | F |
| S3-11 NORMAL PIPELINE | M | P | K | S | S | I | T/Y | F | S | K | M | E | L | L | S | I | W | V | L | L | V | S | S | G | K | S | Y | H | K | S | S | A | D | P | S | R | G | M | F |
| S3-14 FAST PIPELINE | M | L | K | S | S | I | S | L | S | K | M | E | P | P | S | T | W | V | L | F | V | S | A | G | N | S | D | H | K | S | S | S | D | P | I | R | G | M | S |
| S3-14 NORMAL PIPELINE | M | L | K | S | S | I | S | L | S | K | M | E | P | P | S | T | W | V | L | F | V | S | A | G | N | S | D | Q | K | S | S | S | D | P | I | R | G | M | S |
| NGS Sample Code | Running Time (s) | |
|---|---|---|
| Fast Pipeline | Normal Pipeline | |
| B6 | 1778.0 | 5991.3 |
| C5 | 574.3 | 3980.5 |
| F2 | 324.5 | 3924.1 |
| F4 | 286.4 | 3539.5 |
| S3 | 3060.1 | 6521.0 |
| S9 | 1036.8 | 4755.5 |
| S10 | 537.5 | 3747.3 |
| S15 | 848.9 | 4356.8 |
| S3-1 | 552.2 | 4864.7 |
| S3-4 | 256.3 | 4461.3 |
| S3-5 | 1427.8 | 6377.2 |
| S3-7 | 330.6 | 4416.8 |
| S3-8 | 489.6 | 4824.5 |
| S3-9 | 486.5 | 4752.9 |
| S3-11 | 879.6 | 5394.5 |
| S3-14 | 57.6 | 4190.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afiahayati; Bernard, S.; Gunadi; Wibawa, H.; Hakim, M.S.; Marcellus; Parikesit, A.A.; Dewa, C.K.; Sakakibara, Y. A Comparison of Bioinformatics Pipelines for Enrichment Illumina Next Generation Sequencing Systems in Detecting SARS-CoV-2 Virus Strains. Genes 2022, 13, 1330. https://doi.org/10.3390/genes13081330
Afiahayati, Bernard S, Gunadi, Wibawa H, Hakim MS, Marcellus, Parikesit AA, Dewa CK, Sakakibara Y. A Comparison of Bioinformatics Pipelines for Enrichment Illumina Next Generation Sequencing Systems in Detecting SARS-CoV-2 Virus Strains. Genes. 2022; 13(8):1330. https://doi.org/10.3390/genes13081330
Chicago/Turabian StyleAfiahayati, Stefanus Bernard, Gunadi, Hendra Wibawa, Mohamad Saifudin Hakim, Marcellus, Arli Aditya Parikesit, Chandra Kusuma Dewa, and Yasubumi Sakakibara. 2022. "A Comparison of Bioinformatics Pipelines for Enrichment Illumina Next Generation Sequencing Systems in Detecting SARS-CoV-2 Virus Strains" Genes 13, no. 8: 1330. https://doi.org/10.3390/genes13081330
APA StyleAfiahayati, Bernard, S., Gunadi, Wibawa, H., Hakim, M. S., Marcellus, Parikesit, A. A., Dewa, C. K., & Sakakibara, Y. (2022). A Comparison of Bioinformatics Pipelines for Enrichment Illumina Next Generation Sequencing Systems in Detecting SARS-CoV-2 Virus Strains. Genes, 13(8), 1330. https://doi.org/10.3390/genes13081330