
CRISPR DNA Base Editing Strategies for Treating Retinitis Pigmentosa Caused by Mutations in Rhodopsin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
- variant type into insertions/deletions/duplications (indels, not editable with base editors) and SNVs (possibly editable with base editors);
- predicted variant consequence into nonsense, missense, synonymous, splice site (+/− 2 nucleotides from exon-intron boundaries), intronic, 3′ and 5′ untranslated region (UTR), stop loss and start loss variants; and by
- clinical interpretation of the variant pathogenicity into pathogenic/likely pathogenic, variant of unknown clinical significance (VUS), conflicting interpretations of pathogenicity and benign/likely benign variants.
3. Results
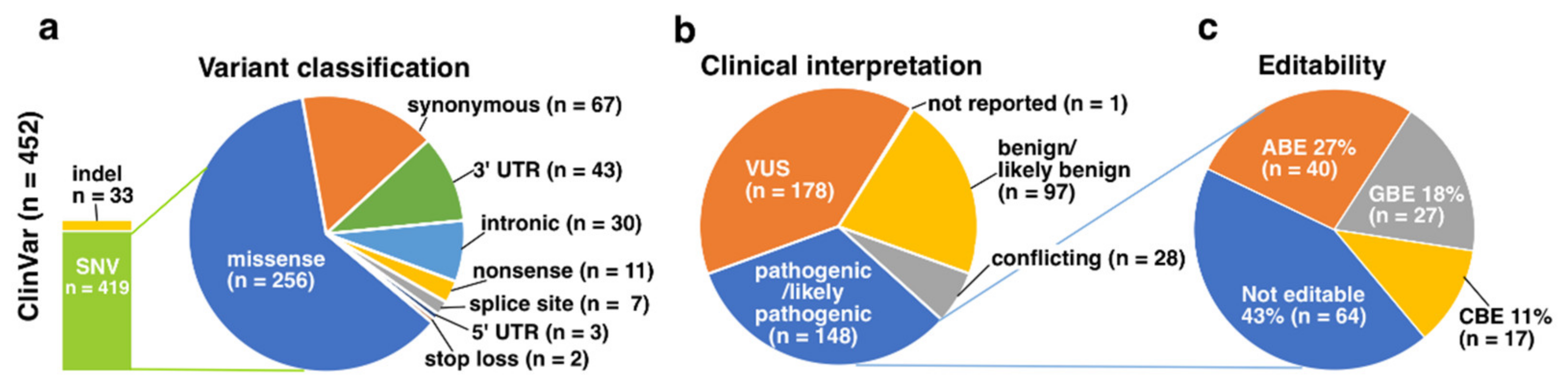
3.1. LOVD
3.2. ClinVar
3.3. gnomAD
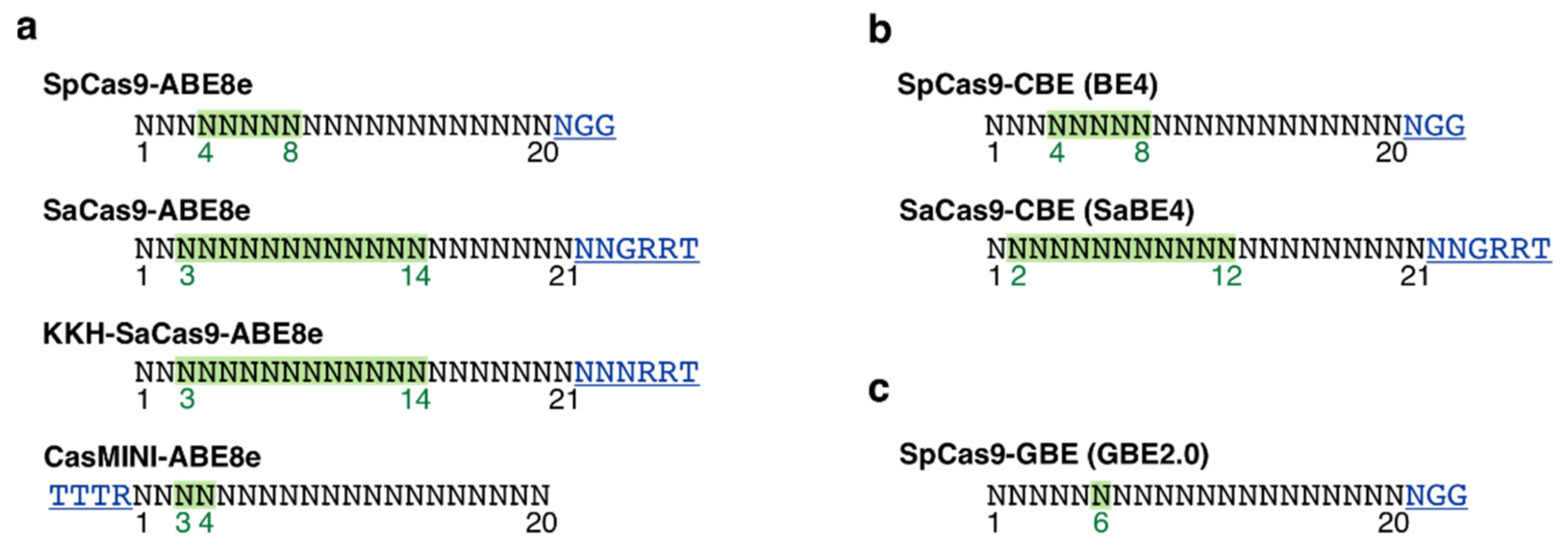
3.4. PAM Site Screening and Guide RNA Design
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Rossiter, B.J.F.; Greenberg, J.; Christoffels, A.; Hide, W. Data services and software for identifying genes and mutations causing retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1998, 39, S295. [Google Scholar]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Cohen, G.B.; Oprian, D.D. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature 1994, 367, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.C.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81. [Google Scholar] [CrossRef]
- Kartasasmita, A.; Fujiki, K.; Iskandar, E.; Sovani, I.; Fujimaki, T.; Murakami, A. A novel nonsense mutation in rhodopsin gene in two Indonesian families with autosomal recessive retinitis pigmentosa. Ophthalmic. Genet. 2011, 32, 57–63. [Google Scholar] [CrossRef]
- Kumaramanickavel, G.; Maw, M.; Denton, M.J.; John, S.; Srikumari, C.R.; Orth, U.; Oehlmann, R.; Gal, A. Missense rhodopsin mutation in a family with recessive RP. Nat. Genet. 1994, 8, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Xiao, X.; Li, S.; Sun, W.; Yi, Z.; Wang, P.; Zhang, Q. Spectrum-frequency and genotype-phenotype analysis of rhodopsin variants. Exp. Eye Res. 2021, 203, 108405. [Google Scholar] [CrossRef]
- Sudharsan, R.; Beltran, W.A. Progress in Gene Therapy for Rhodopsin Autosomal Dominant Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2019, 1185, 113–118. [Google Scholar] [CrossRef]
- Massengill, M.T.; Lewin, A.S. Gene Therapy for Rhodopsin-associated Autosomal Dominant Retinitis Pigmentosa. Int. Ophthalmol. Clin. 2021, 61, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Orlans, H.O.; McClements, M.E.; Barnard, A.R.; Martinez-Fernandez de la Camara, C.; MacLaren, R.E. Mirtron-mediated RNA knockdown/replacement therapy for the treatment of dominant retinitis pigmentosa. Nat. Commun. 2021, 12, 4934. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Tsai, Y.T.; Huang, I.W.; Cheng, C.H.; Hsu, C.W.; Cui, X.; Ryu, J.; Quinn, P.M.J.; Caruso, S.M.; Lin, C.S.; et al. CRISPR genome surgery in a novel humanized model for autosomal dominant retinitis pigmentosa. Mol. Ther. 2022, 30, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Fokkema, I.; Kroon, M.; Lopez Hernandez, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 2021, 81, 4333–4345.e4. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Zhao, D.; Li, S.; Zhang, Z.; Bi, C.; Zhang, X. Reconstructed glycosylase base editors GBE2.0 with enhanced C-to-G base editing efficiency and purity. Mol. Ther. 2022, 30, 2452–2463. [Google Scholar] [CrossRef]
- Fernandez-San Jose, P.; Blanco-Kelly, F.; Corton, M.; Trujillo-Tiebas, M.J.; Gimenez, A.; Avila-Fernandez, A.; Garcia-Sandoval, B.; Lopez-Molina, M.I.; Hernan, I.; Carballo, M.; et al. Prevalence of Rhodopsin mutations in autosomal dominant Retinitis Pigmentosa in Spain: Clinical and analytical review in 200 families. Acta Ophthalmol. 2015, 93, e38–e44. [Google Scholar] [CrossRef]
- Audo, I.; Manes, G.; Mohand-Said, S.; Friedrich, A.; Lancelot, M.E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xiao, X.; Wang, P.; Guo, X.; Zhang, Q. Mutation spectrum and frequency of the RHO gene in 248 Chinese families with retinitis pigmentosa. Biochem. Biophys. Res. Commun. 2010, 401, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Bellingrath, J.S.; McClements, M.E.; Kaukonen, M.; Fischer, M.D.; MacLaren, R.E. In Silico Analysis of Pathogenic CRB1 Single Nucleotide Variants and Their Amenability to Base Editing as a Potential Lead for Therapeutic Intervention. Genes 2021, 12, 1908. [Google Scholar] [CrossRef] [PubMed]
- Piotter, E.; McClements, M.E.; MacLaren, R.E. The Scope of Pathogenic ABCA4 Mutations Targetable by CRISPR DNA Base Editing Systems-A Systematic Review. Front. Genet. 2021, 12, 814131. [Google Scholar] [CrossRef]
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA--occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [Green Version]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [Green Version]
- Berns, K.I.; Giraud, C. Biology of adeno-associated virus. Curr. Top. Microbiol. Immunol. 1996, 218, 1–23. [Google Scholar] [CrossRef]
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Charbel Issa, P.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4(−/−) Mice. Hum. Gene. Ther. 2019, 30, 590–600. [Google Scholar] [CrossRef]
- Suh, S.; Choi, E.H.; Leinonen, H.; Foik, A.T.; Newby, G.A.; Yeh, W.H.; Dong, Z.; Kiser, P.D.; Lyon, D.C.; Liu, D.R.; et al. Restoration of visual function in adult mice with an inherited retinal disease via adenine base editing. Nat. Biomed. Eng. 2021, 5, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gruter, O.; Kostic, C.; Crippa, S.V.; Perez, M.T.; Zografos, L.; Schorderet, D.F.; Munier, F.L.; Arsenijevic, Y. Lentiviral vector-mediated gene transfer in adult mouse photoreceptors is impaired by the presence of a physical barrier. Gene. Ther. 2005, 12, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Barnea-Cramer, A.O.; Singh, M.; Fischer, D.; De Silva, S.; McClements, M.E.; Barnard, A.R.; MacLaren, R.E. Repair of Retinal Degeneration following Ex Vivo Minicircle DNA Gene Therapy and Transplantation of Corrected Photoreceptor Progenitors. Mol. Ther. 2020, 28, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Trigueros, S.; Domenech, E.B.; Toulis, V.; Marfany, G. In Vitro Gene Delivery in Retinal Pigment Epithelium Cells by Plasmid DNA-Wrapped Gold Nanoparticles. Genes 2019, 10, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-Cas Phi from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Gupta, A.; Bednarski, C.; Gehrig-Giannini, S.; Richter, F.; Pitzler, C.; Gamalinda, M.; Galonska, C.; Takeuchi, R.; Wang, K.; et al. Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases. Nat. Commun. 2021, 12, 4219. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Liu, C.-H.; Wolf, P.; Dong, R.; Huang, Y.; Tabbaa, D.; Marco, E.; Duke, B.; Pinilla, A.; Pant, A.; D’Souza, R.; et al. A Mutation-Independent CRISPR/Cas9-based ‘Knockout and Replace’ Strategy to Treat Rhodopsin-Associated Autosomal Dominant Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2022, 63, 3474. [Google Scholar]
- Tan, E.; Wang, Q.; Quiambao, A.B.; Xu, X.; Qtaishat, N.M.; Peachey, N.S.; Lem, J.; Fliesler, S.J.; Pepperberg, D.R.; Naash, M.I.; et al. The relationship between opsin overexpression and photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 589–600. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaukonen, M.; McClements, M.E.; MacLaren, R.E. CRISPR DNA Base Editing Strategies for Treating Retinitis Pigmentosa Caused by Mutations in Rhodopsin. Genes 2022, 13, 1327. https://doi.org/10.3390/genes13081327
Kaukonen M, McClements ME, MacLaren RE. CRISPR DNA Base Editing Strategies for Treating Retinitis Pigmentosa Caused by Mutations in Rhodopsin. Genes. 2022; 13(8):1327. https://doi.org/10.3390/genes13081327
Chicago/Turabian StyleKaukonen, Maria, Michelle E. McClements, and Robert E. MacLaren. 2022. "CRISPR DNA Base Editing Strategies for Treating Retinitis Pigmentosa Caused by Mutations in Rhodopsin" Genes 13, no. 8: 1327. https://doi.org/10.3390/genes13081327
APA StyleKaukonen, M., McClements, M. E., & MacLaren, R. E. (2022). CRISPR DNA Base Editing Strategies for Treating Retinitis Pigmentosa Caused by Mutations in Rhodopsin. Genes, 13(8), 1327. https://doi.org/10.3390/genes13081327