
Challenges in Diagnosing Primary Ciliary Dyskinesia in a Brazilian Tertiary Hospital

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, A.J.; Davis, S.D.; Polineni, D.; Manion, M.; Rosenfeld, M.; Dell, S.D.; Chilvers, M.A.; Ferkol, T.W.; Zariwala, M.A.; Sagel, S.D.; et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 197, e24–e39. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef]
- Lucas, J.S.; Davis, S.D.; Omran, H.; Shoemark, A. Review Primary ciliary dyskinesia in the genomics age. Lancet Respir. 2020, 8, 202–216. [Google Scholar] [CrossRef]
- Goutaki, M.; Meier, A.B.; Halbeisen, F.; Lucas, J.; Dell, S.; Maurer, E.; Casaulta, C.; Jurca, M.; Spycher, B.; Kuehni, C.E. Clinical manifestations in primary ciliary dyskinesia: Systematic review and meta-analysis. Eur. Respir. J. 2016, 48, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Fokkens, W.J.; Lund, V.J.; Hopkins, C.; Hellings, P.W.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J.; Alobid, I.; et al. European position paper on rhinosinusitis and nasal polyps. Rhinology 2020, 58 (Suppl. S29), 1–464. [Google Scholar] [CrossRef]
- Afzelius, B.A. A Human Syndrome Caused by Immotile Cilia. Science 1976, 193, 317–319. [Google Scholar] [CrossRef]
- Lucas, J.S.; Leigh, M.W. Diagnosis of primary ciliary dyskinesia: Searching for a gold standard. Eur. Respir. J. 2014, 44, 1418–1422. [Google Scholar] [CrossRef] [Green Version]
- Shoemark, A.; Dell, S.; Shapiro, A.; Lucas, J.S. ERS and ATS diagnostic guidelines for primary ciliary dyskinesia: Similarities and differences in approach to diagnosis. Eur. Respir. J. 2019, 54, 1901066. [Google Scholar] [CrossRef]
- Amirav, I.; Lavie, M. Reply to Shoemark et al. And to Shapiro et al. Am. J. Respir. Crit. Care Med. 2020, 201, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.W.; Ferkol, T.W.; Davis, S.D.; Lee, H.-S.; Rosenfeld, M.; Dell, S.; Sagel, S.D.; Milla, C.; Olivier, K.N.; Sullivan, K.M.; et al. Clinical Features and Associated Likelihood of Primary Ciliary Dyskinesia in Children and Adolescents. Ann. Am. Thorac. Soc. 2016, 13, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, L.; Dimitrov, B.D.; Kuehni, C.E.; Hogg, C.; Carroll, M.; Evans, H.J.; Goutaki, M.; Harris, A.; Packham, S.; Walker, W.T.; et al. PICADAR: A diagnostic predictive tool for primary ciliary dyskinesia. Eur. Respir. J. 2016, 47, 1103–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinů, V.; Bořek-Dohalská, L.; Varényiová, Ž.; Uhlík, J.; Čapek, V.; Pohunek, P.; Koucký, V. Evaluation of a Clinical Index as a Predictive Tool for Primary Ciliary Dyskinesia. Diagnostics 2021, 11, 1088. [Google Scholar] [CrossRef]
- Rubbo, B.; Shoemark, A.; Jackson, C.L.; Hirst, R.; Thompson, J.; Hayes, J.; Frost, E.; Copeland, F.; Hogg, C.; O’Callaghan, C.; et al. Accuracy of High-Speed Video Analysis to Diagnose Primary Ciliary Dyskinesia. Chest 2019, 155, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Hogg, C.; Bush, A. CON: Primary Ciliary Dyskinesia diagnosis: Genes are all you need! Paediatr. Respir. Rev. 2021, 37, 34–36. [Google Scholar] [CrossRef]
- Shirlow, R.; Fitzgerald, D.A. PRO: Primary Ciliary Dyskinesia: Genes are all you need! Paediatr. Respir. Rev. 2021, 37, 32–33. [Google Scholar] [CrossRef]
- Goutaki, M.; Maurer, E.; Halbeisen, F.; Amirav, I.; Barbato, A.; Behan, L.; Boon, M.; Casaulta, C.; Clement, A.; Crowley, S.; et al. The international primary ciliary dyskinesia cohort (iPCD Cohort): Methods and first results. Eur. Respir. J. 2017, 49, 1601181. [Google Scholar] [CrossRef] [Green Version]
- Rumman, N.; Jackson, C.; Collins, S.; Goggin, P.; Coles, J.; Lucas, J. Diagnosis of primary ciliary dyskinesia: Potential options for resource-limited countries. Eur. Respir. Rev. 2017, 26, 160058. [Google Scholar] [CrossRef]
- Piatti, G.; De Santi, M.M.; Farolfi, A.; Zuccotti, G.V.; D’Auria, E.; Patria, M.F.; Torretta, S.; Consonni, D.; Ambrosetti, U. Exacerbations and Pseudomonas aeruginosa colonization are associated with altered lung structure and function in primary ciliary dyskinesia. BMC Pediatr. 2020, 20, 158. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.A.K.; Marson, F.A.L.; Athanazio, R.A.; Nakagawa, N.K.; Macchione, M.; Loges, N.T.; Omran, H.; Rached, S.Z.; Bertuzzo, C.S.; Stelmach, R.; et al. Severe pulmonary disease in an adult primary ciliary dyskinesia population in Brazil. Sci. Rep. 2019, 9, 8693. [Google Scholar] [CrossRef]
- Kuehni, C.E.; Lucas, J.S. Diagnosis of primary ciliary dyskinesia: Summary of the ERS Task Force report. Breathe 2017, 13, 166–178. [Google Scholar] [CrossRef]
- Shoemark, A.; Boon, M.; Brochhausen, C.; Bukowy-Bieryllo, Z.; De Santi, M.M.; Goggin, P.; Griffin, P.; Hegele, R.G.; Hirst, R.A.; Leigh, M.W.; et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM Criteria). Eur. Respir. J. 2020, 55, 1900725. [Google Scholar] [CrossRef]
- Olm, M.A.K.; Caldini, E.G.; Mauad, T. Diagnosis of primary ciliary dyskinesia. J. Bras. Pneumol. 2015, 41, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carceller, M.A.; Roig, M.M.; Payá, X.M.; Gimeno, J.C. Discinesia ciliar primaria. Ciliopatías. Acta Otorrinolaringol. Esp. 2010, 61, 149–159. [Google Scholar]
- Boone, M.; Smits, A.; Cuppens, H.; Jaspers, M.; Proesmans, M.; Dupont, L.J.; Vermeulen, F.L.; Van Daele, S.; Malfroot, A.; Godding, V.; et al. Primary ciliary dyskinesia: Critical evaluation of clinical symptoms and diagnosis in patients with normal and abnormal ultrastructure. Orphanet J. Rare Dis. 2014, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kuehni, C.E.; Lucas, J.S. Toward an earlier diagnosis of primary ciliary dyskinesia which patients should undergo detailed diagnostic testing? Ann. Am. Thorac. Soc. 2016, 13, 1239–1243. [Google Scholar] [CrossRef]
- Hammoudeh, S.; Gadelhak, W.; Janahi, I.A. Primary ciliary dyskinesia among Arabs: Where do we go from here? Paediatr. Respir. Rev. 2019, 29, 19–22. [Google Scholar] [CrossRef]
- Kuehni, C.E.; Frischer, T.; Strippoli, M.-P.; Maurer, E.; Bush, A.; Nielsen, K.G.; Escribano, A.; Lucas, J.; Yiallouros, P.; Omran, H.; et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur. Respir. J. 2010, 36, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Goutaki, M.; Halbeisen, F.; Barbato, A.; Crowley, S.; Harris, A.; Hirst, R.; Karadag, B.; Martinu, V.; Morgan, L.; O’Callaghan, C.; et al. Late Diagnosis of Infants with PCD and Neonatal Respiratory Distress. J. Clin. Med. 2020, 9, 2871. [Google Scholar] [CrossRef]
- Behan, L.; Galvin, A.D.; Rubbo, B.; Masefield, S.C.; Copeland, F.; Manion, M.; Rindlisbacher, B.; Redfern, B.; Lucas, J.S. Diagnosing primary ciliary dyskinesia: An international patient perspective. Eur. Respir. J. 2016, 48, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Behan, L.; Rubbo, B.; Lucas, J.S.; Galvin, A.D. The patient’s experience of primary ciliary dyskinesia: A systematic review. Qual. Life Res. 2017, 26, 2265–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.A.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5Mutations Are a Common Cause of Primary Ciliary Dyskinesia with Outer Dynein Arm Defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in Respiratory Cells from Patients with Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Failly, M.; Bartoloni, L.; Letourneau, A.; Munoz, A.; Falconnet, E.; Rossier, C.; de Santi, M.M.; Santamaria, F.; Sacco, O.; DeLozier-Blanchet, C.D.; et al. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J. Med. Genet. 2009, 46, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Leigh, M.W. Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct. Pathol. 2017, 41, 373–385. [Google Scholar] [CrossRef]
- Knowles, M.R.; Leigh, M.W.; Carson, J.L.; Davis, S.D.; Dell, S.D.; Ferkol, T.W.; Olivier, K.N.; Sagel, S.D.; Rosenfeld, M.; Burns, K.A.; et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 2012, 67, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.; et al. CCDC151 Mutations Cause Primary Ciliary Dyskinesia by Disruption of the Outer Dynein Arm Docking Complex Formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 Mutations Cause Primary Ciliary Dyskinesia with Randomization of Left/Right Body Asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Blanchon, S.; Legendre, M.; Copin, B.; Duquesnoy, P.; Montantin, G.; Kott, E.; Dastot, F.; Jeanson, L.; Cachanado, M.; Rousseau, A.; et al. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J. Med. Genet. 2012, 49, 410–416. [Google Scholar] [CrossRef]
- Shoemark, A.; Ives, A.; Becker-Heck, A.; Burgoyne, T.; Dixon, M.; Bilton, D.; Wilson, R.; Omran, H.; Hogg, C. Inner Dynein Arm Defects in Primary Ciliary Dyskinesia. J. Genet. Syndr. Gene Ther. 2013, 4, 163. [Google Scholar] [CrossRef] [Green Version]
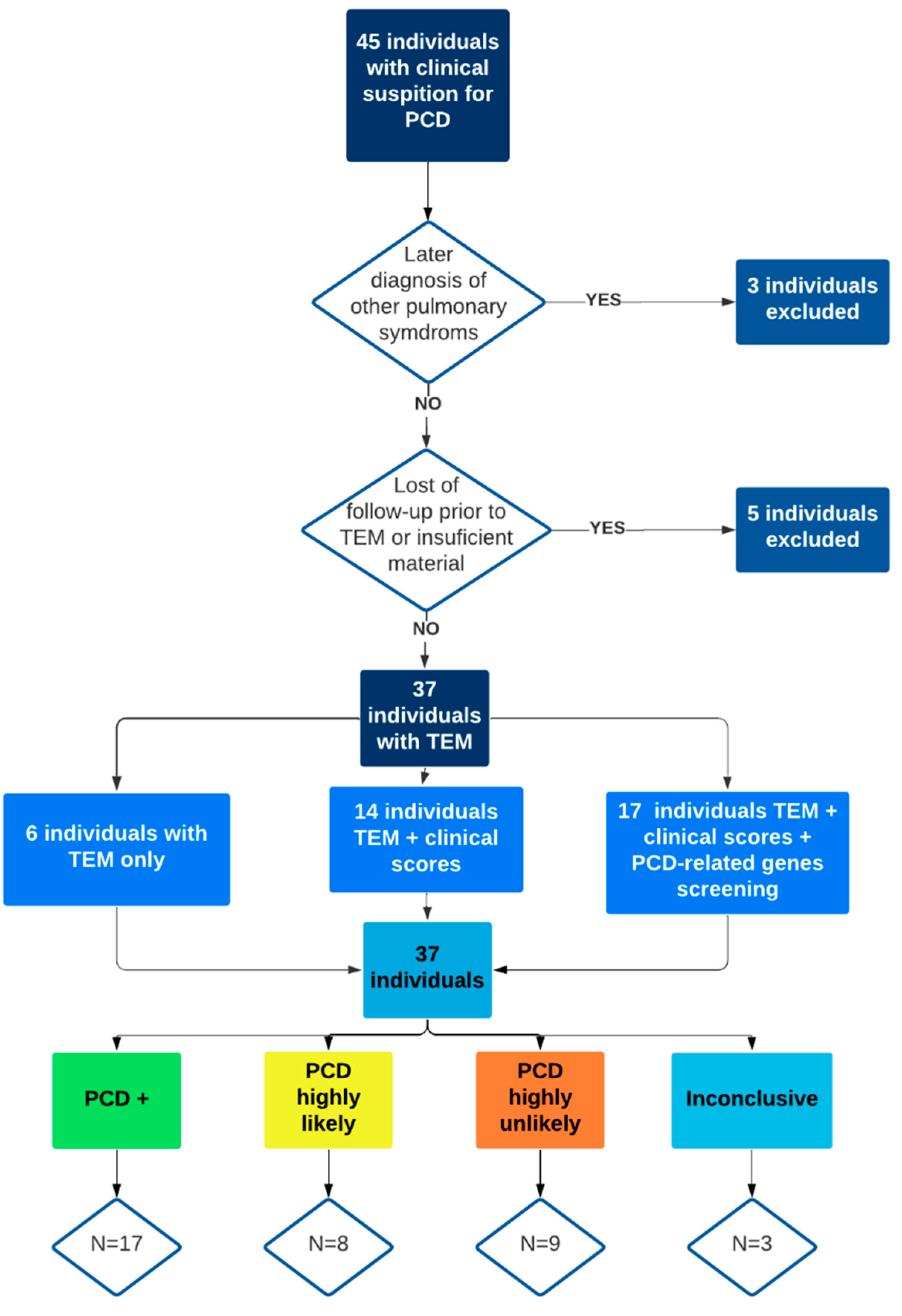

{kind=link}
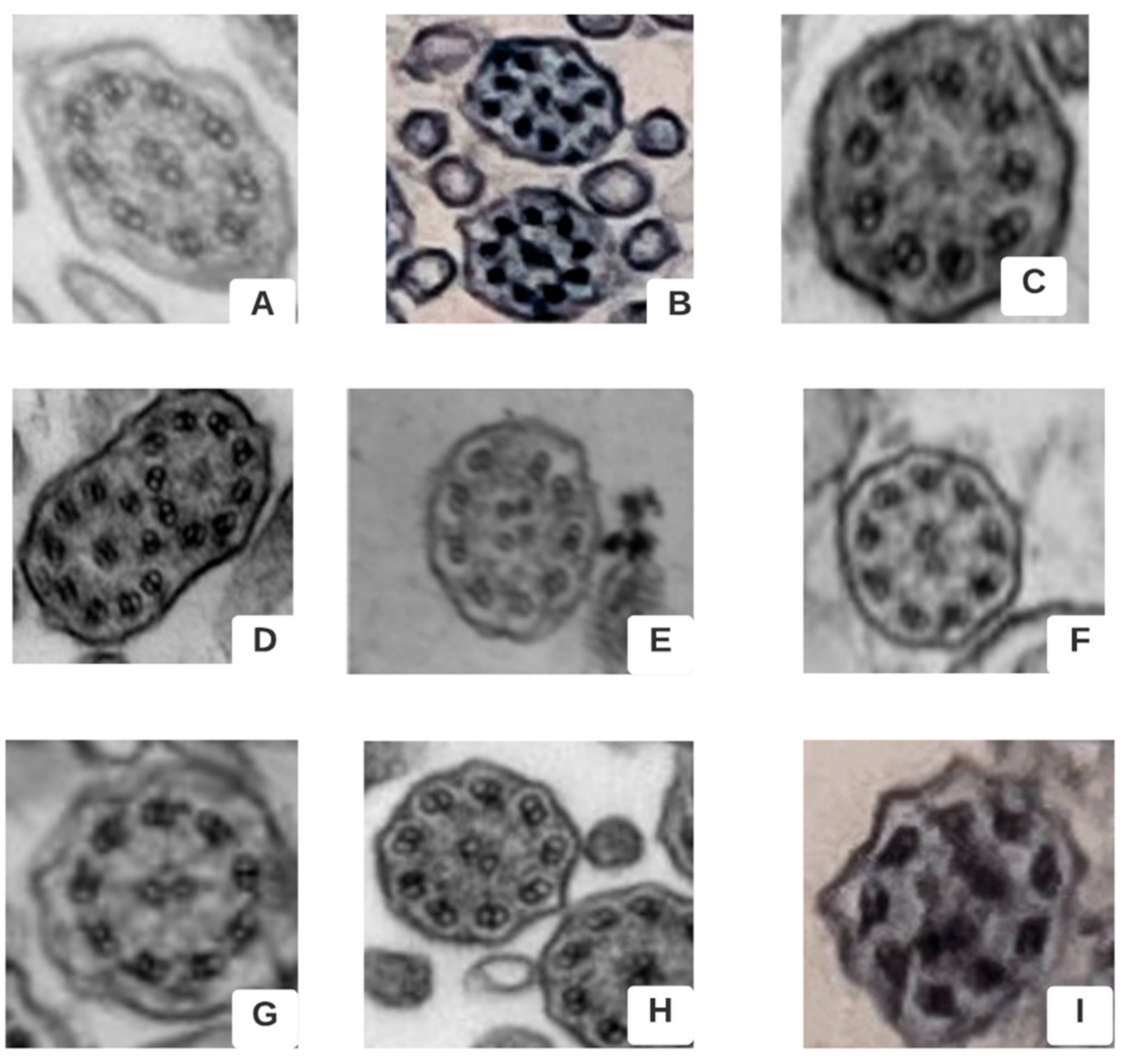
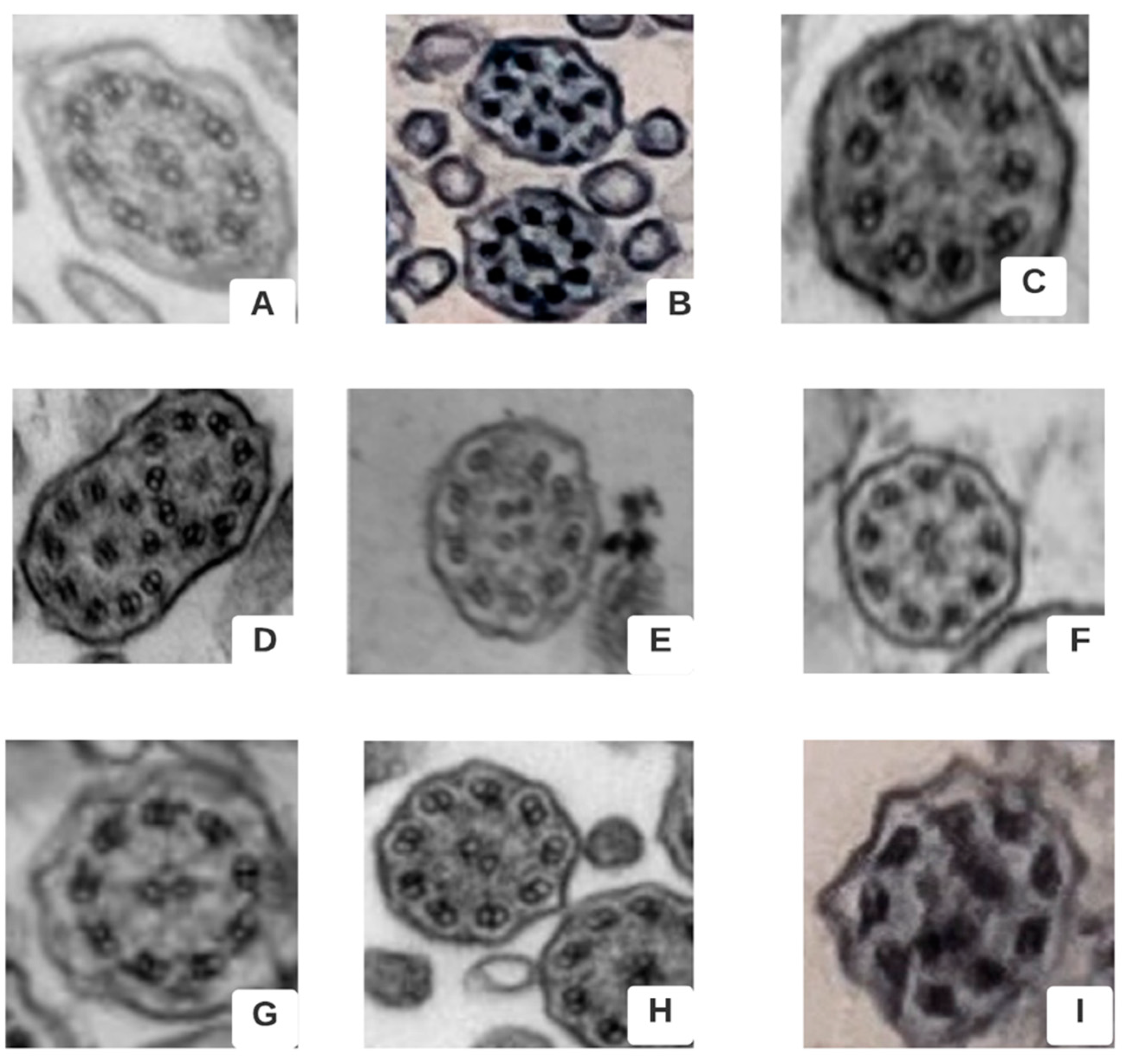{kind=link}
| Case | PCD Diagnosis | Time to Diagnosis * | CRS | NP | CR | Asthma | Bronchiectasis | LD | RP | CO | FD | Consanguinity | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low clinical suspicion | 1 | PCD+ | 4 | - | - | + | - | + | - | - | + | - | - |
| 2 | PCD highly unlikely | - | - | + | + | - | - | + | + | - | - | ||
| 3 | Inconclusive | - | - | + | - | - | - | + | - | - | - | ||
| 4 | PCD+ | 0 | - | - | + | + | - | - | + | - | - | - | |
| 5 | PCD highly unlikely | - | - | - | - | + | - | + | - | - | - | ||
| 6 | PCD highly unlikely | + | + | + | + | - | - | - | - | - | - | ||
| 7 | PCD highly unlikely | - | - | + | + | + | - | + | - | - | - | ||
| 8 | PCD highly unlikely | - | - | + | + | - | - | + | - | - | - | ||
| Moderate clinical suspicion | 9 | PCD highly unlikely | - | - | + | + | + | - | + | + | - | - | |
| 10 | PCD highly likely | + | + | - | - | + | - | + | + | - | + | ||
| 11 | PCD highly likely | + | - | + | + | + | - | + | + | - | - | ||
| 12 | PCD highly unlikely | - | - | + | + | + | - | + | + | - | - | ||
| 13 | PCD+ | 3 | + | - | - | - | + | - | + | + | - | - | |
| 14 | PCD+ | 1 | + | - | + | + | + | - | + | + | - | - | |
| 15 | PCD+ | 1 | + | - | - | - | + | - | + | + | - | - | |
| 16 | PCD highly unlikely | + | + | - | + | + | - | - | - | - | - | ||
| 17 | PCD highly likely | - | - | + | + | + | - | - | - | - | - | ||
| 18 | PCD+ | 2 | + | + | - | - | + | - | + | - | - | - | |
| 19 | PCD highly likely | - | - | - | + | - | - | + | - | - | - | ||
| 20 | PCD highly unlikely | - | - | + | + | + | - | + | + | - | - | ||
| 21 | PCD highly likely | + | + | - | - | + | - | + | - | - | - | ||
| 22 | PCD+ | 5 | - | - | + | - | + | - | + | - | - | - | |
| 23 | Inconclusive | - | - | - | - | - | - | + | - | - | - | ||
| 24 | Inconclusive | + | - | + | - | + | - | + | + | - | - | ||
| High clinical suspicion | 25 | PCD+ | 14 | + | - | - | - | + | - | - | - | + | - |
| 26 | PCD highly likely | + | + | - | - | + | + | + | - | + | - | ||
| 27 | PCD+ | 7 | + | + | - | - | + | + | - | + | - | - | |
| 28 | PCD+ | 12 | + | - | - | - | + | + | + | - | - | - | |
| 29 | PCD+ | 19 | + | - | - | - | + | + | - | - | - | - | |
| 30 | PCD+ | 24 | - | - | + | + | + | + | + | + | - | + | |
| 31 | PCD+ | 1 | + | - | - | + | + | - | - | - | + | - | |
| 32 | PCD highly likely | + | - | - | - | + | + | + | + | - | - | ||
| 33 | PCD highly likely | + | - | - | - | - | + | + | + | - | - | ||
| 34 | PCD+ | 11 | - | - | + | - | - | + | + | + | - | - | |
| 35 | PCD+ | 0 | - | - | - | - | + | + | + | + | - | + | |
| 36 | PCD+ | 2 | + | + | - | - | + | + | + | + | - | - | |
| 37 | PCD+ | 22 | + | - | - | - | + | + | + | - | + | - |
| N | Median Age Suspicion * | Minimum | Maximum | P25 | P75 | |
| Low suspicion | 8 | 13 | 13 | 13 | 13 | 13 |
| Moderate suspicion | 16 | 9 | 2 | 48 | 8 | 12 |
| High suspicion | 13 | 3.5 | 0 | 46 | 1 | 15 |
| Total | 37 | 8 | 0 | 48 | 1 | 13 |
| N | Median age diagnosis ** | Minimum | Maximum | P25 | P75 | |
| Low suspicion | 2 | 15 | 13 | 17 | 13 | 17 |
| Moderate suspicion | 5 | 11 | 7 | 50 | 10 | 13 |
| High suspicion | 10 | 19.5 | 2 | 60 | 9 | 32 |
| Total | 17 | 13 | 2 | 60 | 10 | 25 |
| Case | Sex | Age at Suspicion | TEM a | Genetics | PICADAR ≥ 7 | ATS-CSQ ≥ 2 | PCD Diagnosis | Age at Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| Low clinical suspicion | 1 | F | 13 | Class I | - | - | - | PCD+ | 17 |
| 2 | M | 8 | Normal | N/D | - | + | PCD highly unlikely | ||
| 3 | M | 9 | Class II | N/D | - | - | Inconclusive | ||
| 4 | F | 13 | Class I | N/D | - | + | PCD+ | 13 | |
| 5 | M | 7 | Normal | N/D | N/D | N/D | PCD highly unlikely | ||
| 6 | M | 15 | Normal | N/D | N/D | N/D | PCD highly unlikely | ||
| 7 | M | 10 | Normal | N/D | - | - | PCD highly unlikely | ||
| 8 | M | 15 | Normal | N/D | - | - | PCD highly unlikely | ||
| Moderate clinical suspicion | 9 | F | 16 | Normal | N/D | - | + | PCD highly unlikely | |
| 10 | M | 0 | Class II | N/D | + | + | PCD highly likely | ||
| 11 | F | 9 | Class II | N/D | - | + | PCD highly likely | ||
| 12 | M | 16 | Normal | - | - | + | PCD highly unlikely | ||
| 13 | F | 8 | Class I | + | + | + | PCD+ | 11 | |
| 14 | M | 12 | Class I | N/D | - | - | PCD+ | 13 | |
| 15 | M | 9 | Class I | + | + | + | PCD+ | 10 | |
| 16 | M | 44 | Normal | N/D | N/D | N/D | PCD highly unlikely | ||
| 17 | F | 4 | Class II | N/D | + | + | PCD highly likely | ||
| 18 | M | 48 | Class I | - | - | + | PCD+ | 50 | |
| 19 | M | 1 | Normal | + * | - | - | PCD highly likely | ||
| 20 | F | 15 | Normal | N/D | - | + | PCD highly unlikely | ||
| 21 | M | 21 | Normal | + * | - | + | PCD highly likely | ||
| 22 | M | 2 | Class I | - | - | - | PCD+ | 7 | |
| 23 | F | 2 | Normal | N/D | + | + | Inconclusive | ||
| 24 | F | 3 | Normal | N/D | - | - | Inconclusive | ||
| High clinical suspicion | 25 | M | 46 | Class I | N/D | N/D | N/D | PCD+ | 60 |
| 26 | M | 27 | Class II | N/D | N/D | N/D | PCD highly likely | ||
| 27 | M | 25 | Class I | + | + | + | PCD+ | 32 | |
| 28 | M | 0 | Class I | + | + | + | PCD+ | 12 | |
| 29 | F | 15 | Class I | - | + | + | PCD+ | 34 | |
| 30 | M | 1 | Normal | + | + | + | PCD+ | 25 | |
| 31 | M | 28 | Class I | N/D | N/D | N/D | PCD+ | 28 | |
| 32 | M | 0 | Class II | - | + | + | PCD highly likely | ||
| 33 | F | 8 | Class II | + * | + | + | PCD highly likely | ||
| 34 | M | 6 | Class I | N/D | - | + | PCD+ | 17 | |
| 35 | F | 1 | Class I | - | + | + | PCD+ | 2 | |
| 36 | M | 7 | Class I | + | + | + | PCD+ | 9 | |
| 37 | M | 0 | Class I | + | - | + | PCD+ | 22 |
| Case | Clinical Findings | TEM Findings | Gene | Protein | c.DNA | code | Alleles | Expected Ultrastructural Alterations * |
|---|---|---|---|---|---|---|---|---|
| Patients who received a definitive primary ciliary dyskinesia diagnosis by a conclusive genetic test.a | ||||||||
| 13 | BCT, CRS, CO, RP | IDA + ODA/MD | DNAH11 | p.Cys1597Phe | c.4790G>T | rs72657327 | Hom | Normal ultrastructure/ODA defects |
| CCDC40 | p.Ala83ValfsTer84 and p.Leu872Ter | c.248delC and c.2614delC | Without id and rs775128843 | Het and het | 96 nm axonemal ruler: IDA+MD | |||
| 15 | BCT, CO, CRS, RP | IDA + ODA | DNAH5 | p.Arg4577Ter | c.13729C>T and c.11571-1G>A | Both variants did not have an id | Het and het | ODA defects |
| 27 | BCT, CO, CRS, SI | IDA + ODA | CCDC151 | p.His199ArgfsTer60 | c.583_595dupGCGCAAAACAGAC | rs750658321 | Hom | ODA docker |
| 28 | BCT, CRS, DC, RP | IDA + CCD + MD | CCDC40 | p.Leu872Ter and p.Ala83ValfsTer84 | c.2614delC and c.248delC | rs775128843 and rs397515393 | Het and het | 96 nm axonemal ruler: IDA+MD |
| 30 | AR, Asthma, BCT, CH, CO, RP, SI | - | CCDC151 | p.His199ArgfsTer60 | c.583_595dupGCGCAAAACAGAC | rs750658321 | Hom | ODA docker |
| 36 | BCT, CO, CRS, DC, RP | IDA + ODA + CCD | ARMC4 | p.Gln320SerfsTer44 | c.958delC | Without id | Hom | ODA docker |
| 37 | BCT, CRS, RP, SD, SI | IDA + ODA | DNAI2 | p.Arg263Ter | c.787C>T | rs137852998 | Hom | ODA defects |
| Patients who received an inclusive primary ciliary dyskinesia in the genetic test due the presence of only one pathogenic variant.b | ||||||||
| 19 | Asthma, FH+, RP | IDA | DNAH11 | p.Met1096Ile | c.3288G>A | rs575775297 | Het | Normal ultrastructure/ODA defects |
| 21 | CRS, BCT, FH+, RP | IDA | DNAH11 | p.Met1096Ile | c.3288G>A | rs575775297 | Het | Normal ultrastructure/ODA defects |
| 33 | CO, CRS, RP, SI | IDA + CCD | DNAH5 | p.Arg3885Ter | c.11653C>T | rs756032160 | Het | ODA defects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toro, M.D.C.; Ribeiro, J.D.; Marson, F.A.L.; Ortiz, É.; Toro, A.A.D.C.; Bertuzzo, C.S.; Jones, M.H.; Sakano, E. Challenges in Diagnosing Primary Ciliary Dyskinesia in a Brazilian Tertiary Hospital. Genes 2022, 13, 1252. https://doi.org/10.3390/genes13071252
Toro MDC, Ribeiro JD, Marson FAL, Ortiz É, Toro AADC, Bertuzzo CS, Jones MH, Sakano E. Challenges in Diagnosing Primary Ciliary Dyskinesia in a Brazilian Tertiary Hospital. Genes. 2022; 13(7):1252. https://doi.org/10.3390/genes13071252
Chicago/Turabian StyleToro, Mariana Dalbo Contrera, José Dirceu Ribeiro, Fernando Augusto Lima Marson, Érica Ortiz, Adyléia Aparecida Dalbo Contrera Toro, Carmen Silvia Bertuzzo, Marcus Herbert Jones, and Eulália Sakano. 2022. "Challenges in Diagnosing Primary Ciliary Dyskinesia in a Brazilian Tertiary Hospital" Genes 13, no. 7: 1252. https://doi.org/10.3390/genes13071252
APA StyleToro, M. D. C., Ribeiro, J. D., Marson, F. A. L., Ortiz, É., Toro, A. A. D. C., Bertuzzo, C. S., Jones, M. H., & Sakano, E. (2022). Challenges in Diagnosing Primary Ciliary Dyskinesia in a Brazilian Tertiary Hospital. Genes, 13(7), 1252. https://doi.org/10.3390/genes13071252