
Immune-Related Genomic Schizophrenic Subtyping Identified in DLPFC Transcriptome


Abstract
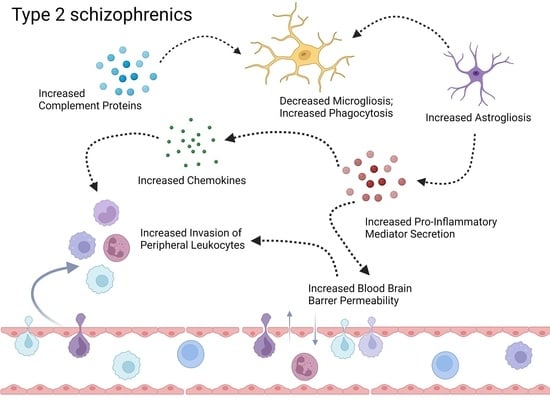
:
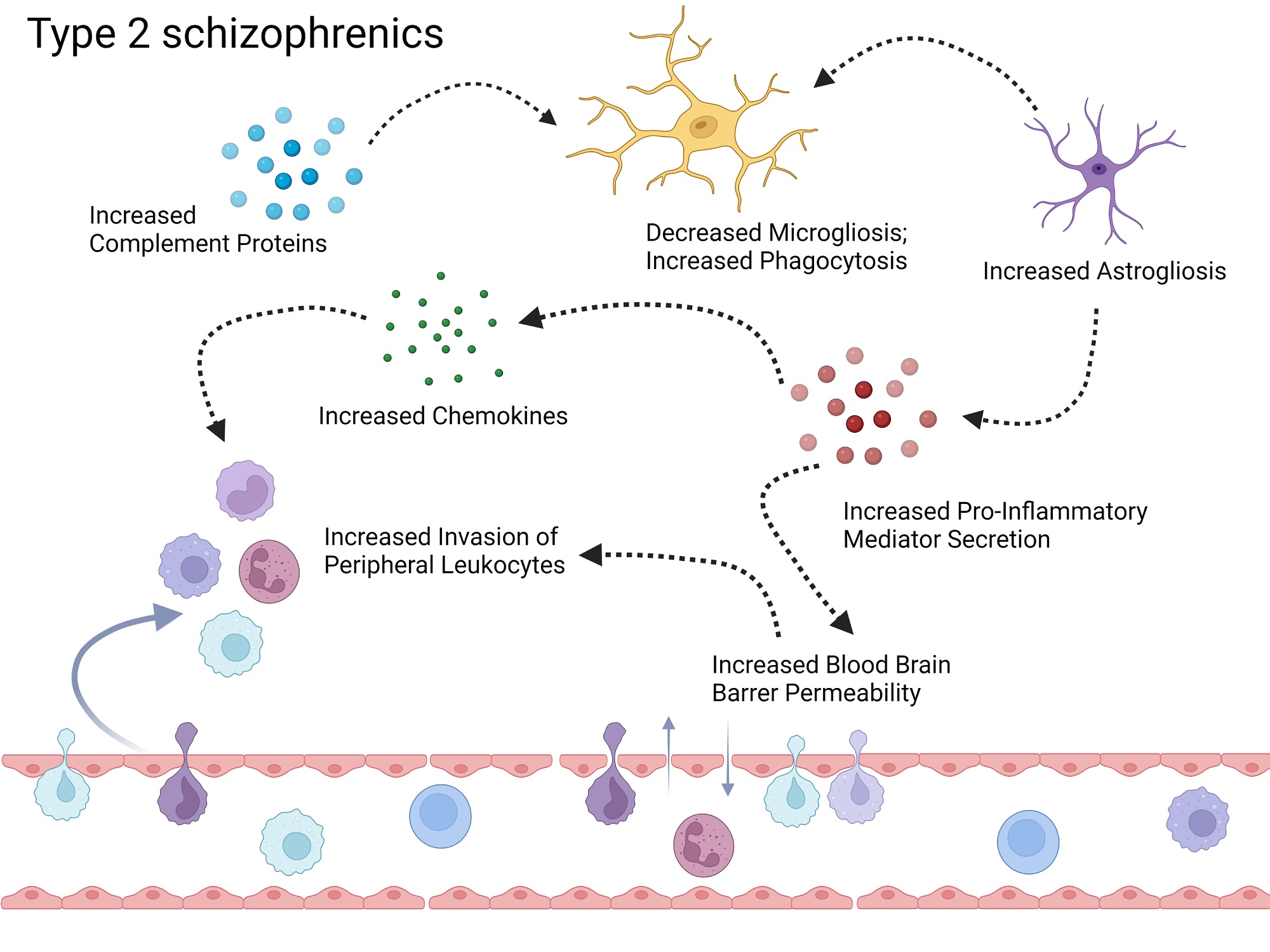{kind=link}
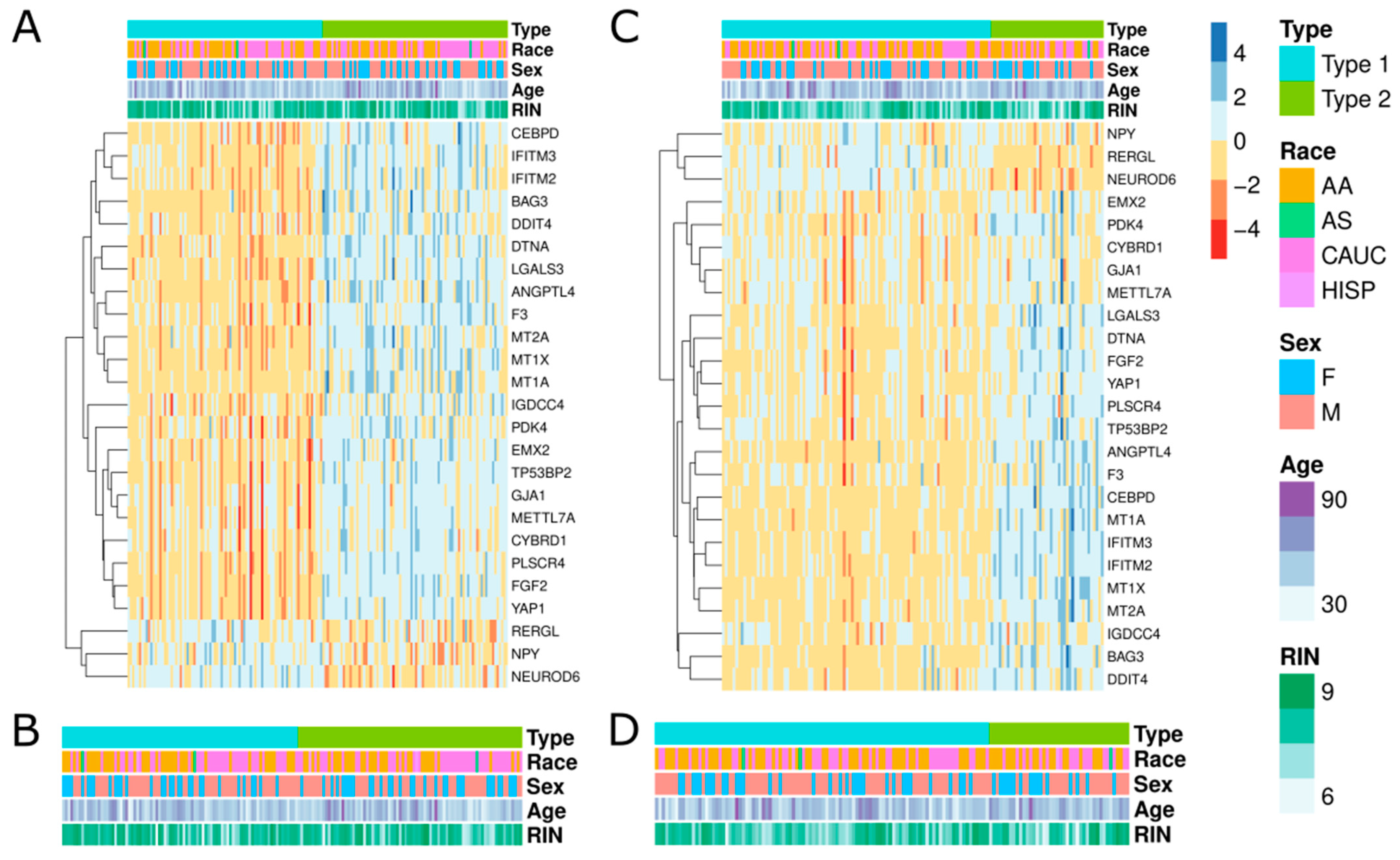{kind=link}
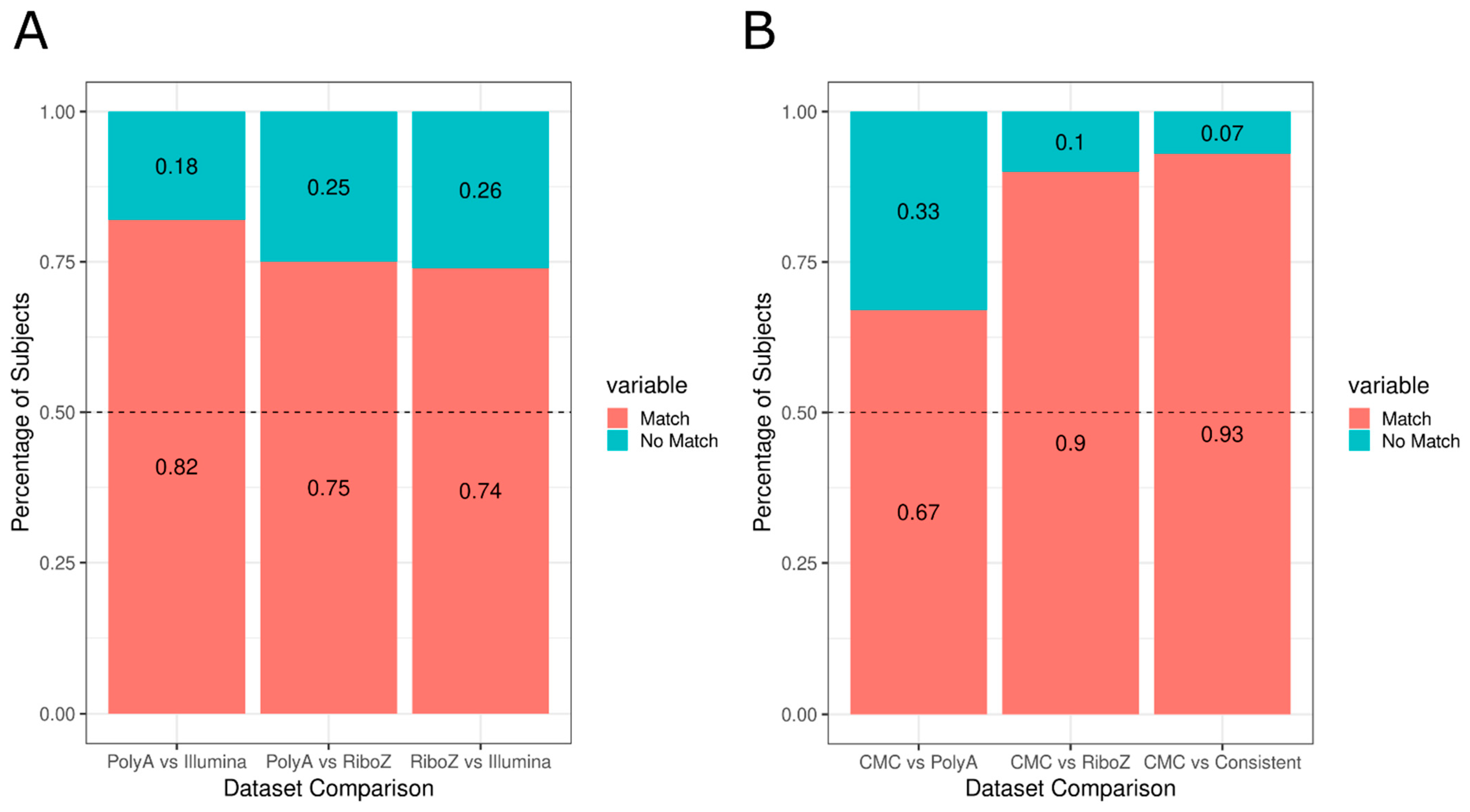{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sources of Data
2.2. Transcriptome Analysis
2.3. Cluster Analysis
2.4. Type 1 vs. Type 2 Differentiation
2.5. Gene List Creation
3. Results
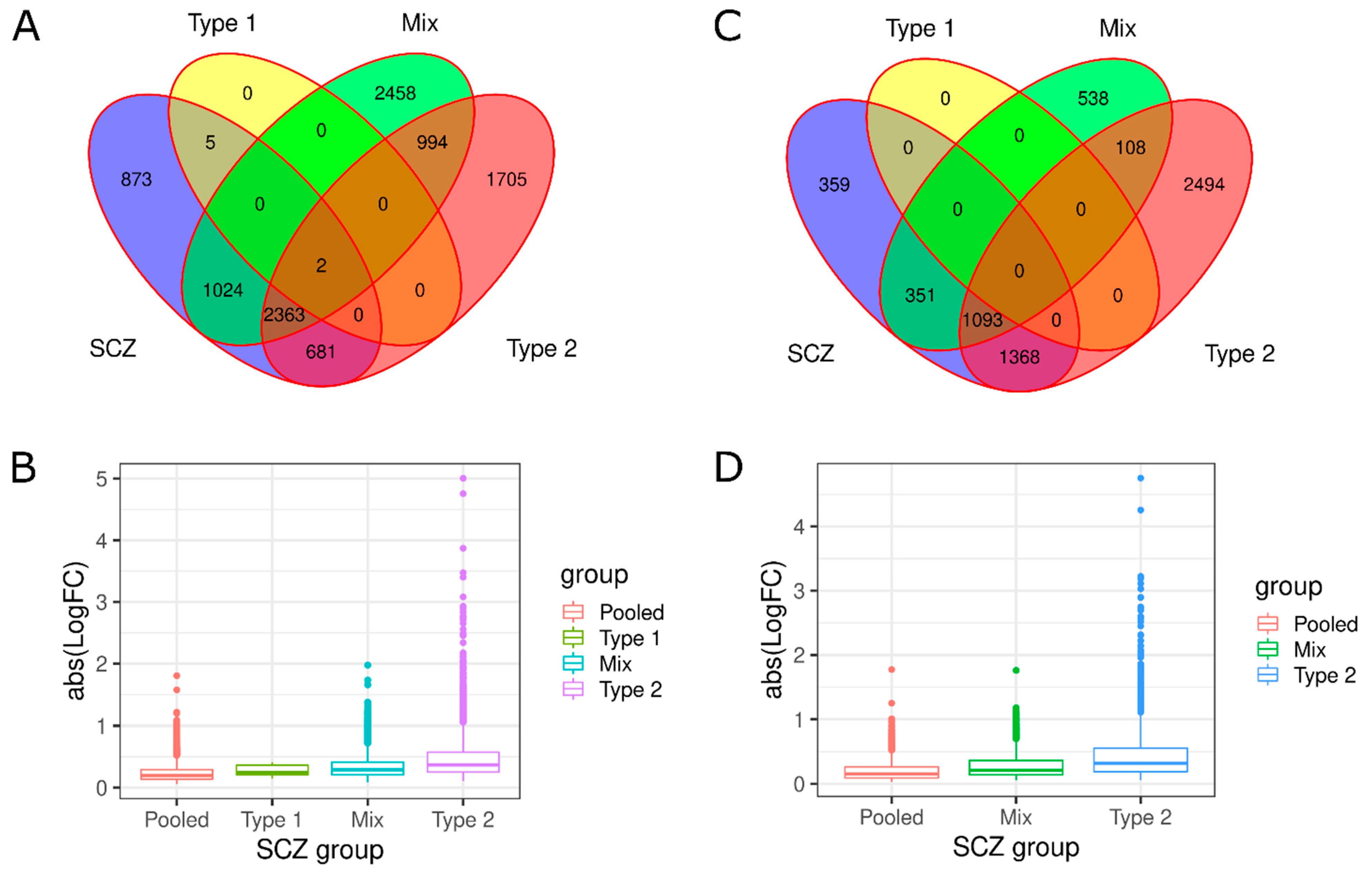
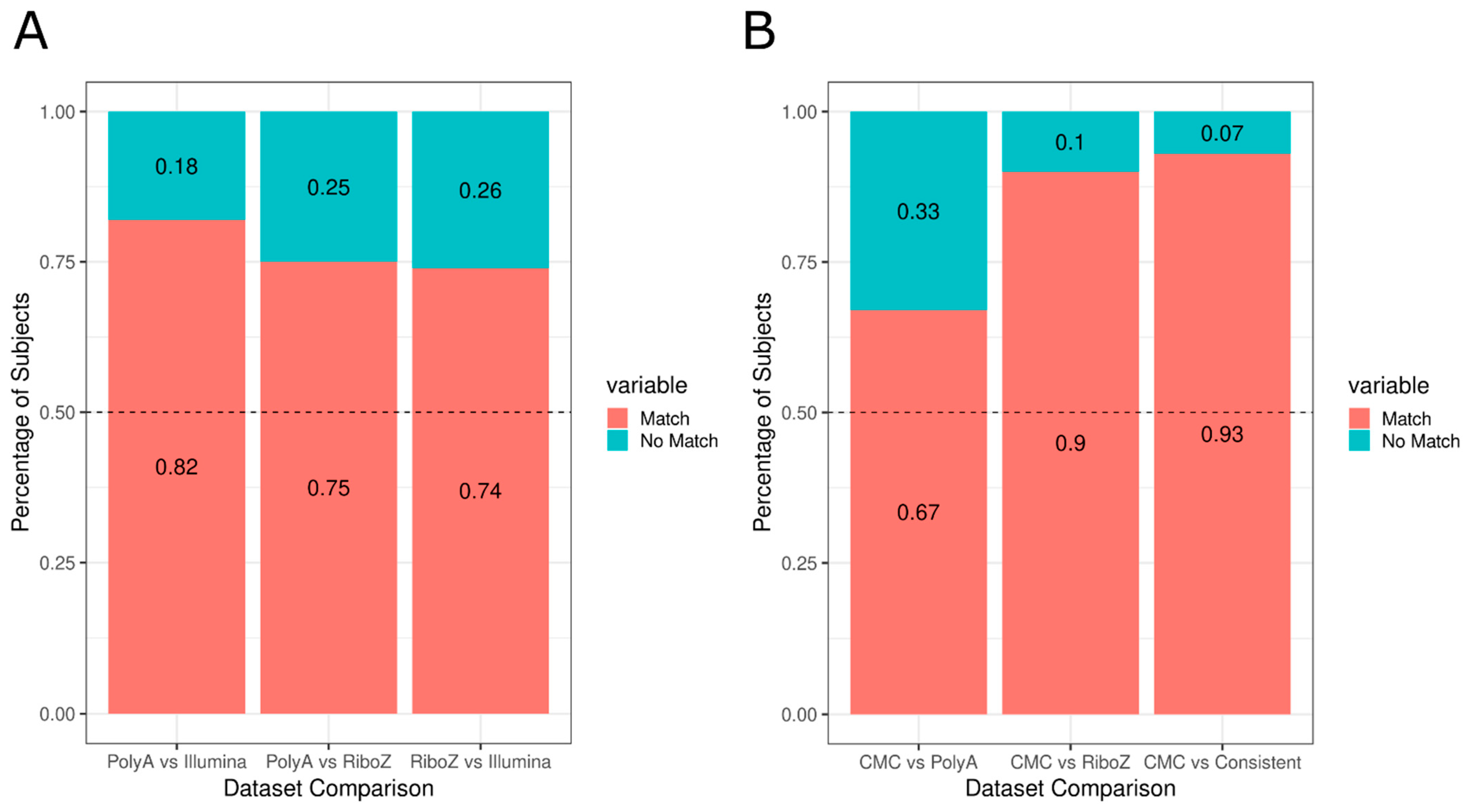
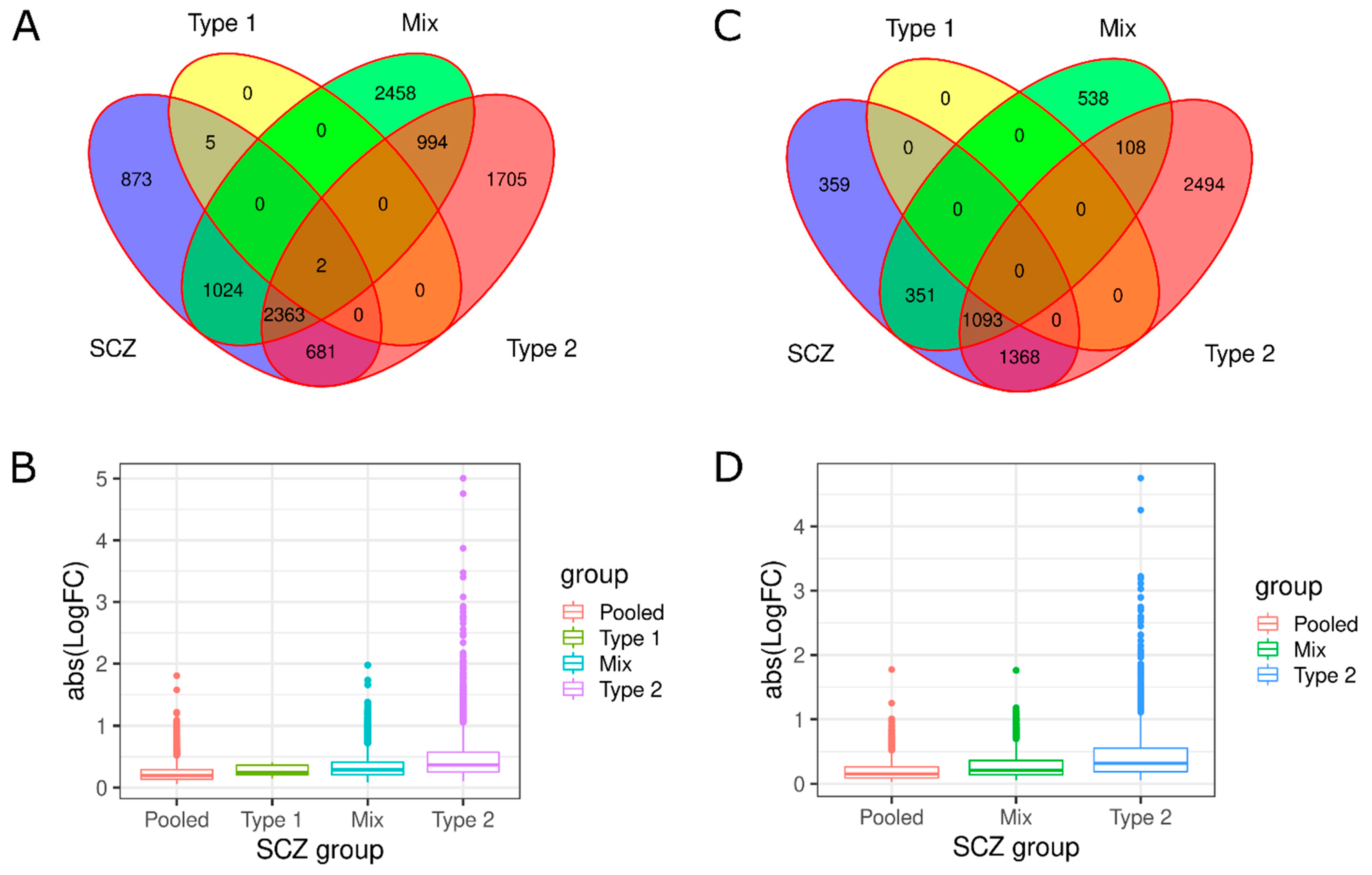
3.1. Investigation of Replicabilty of Two Schizophrenic Subtypes
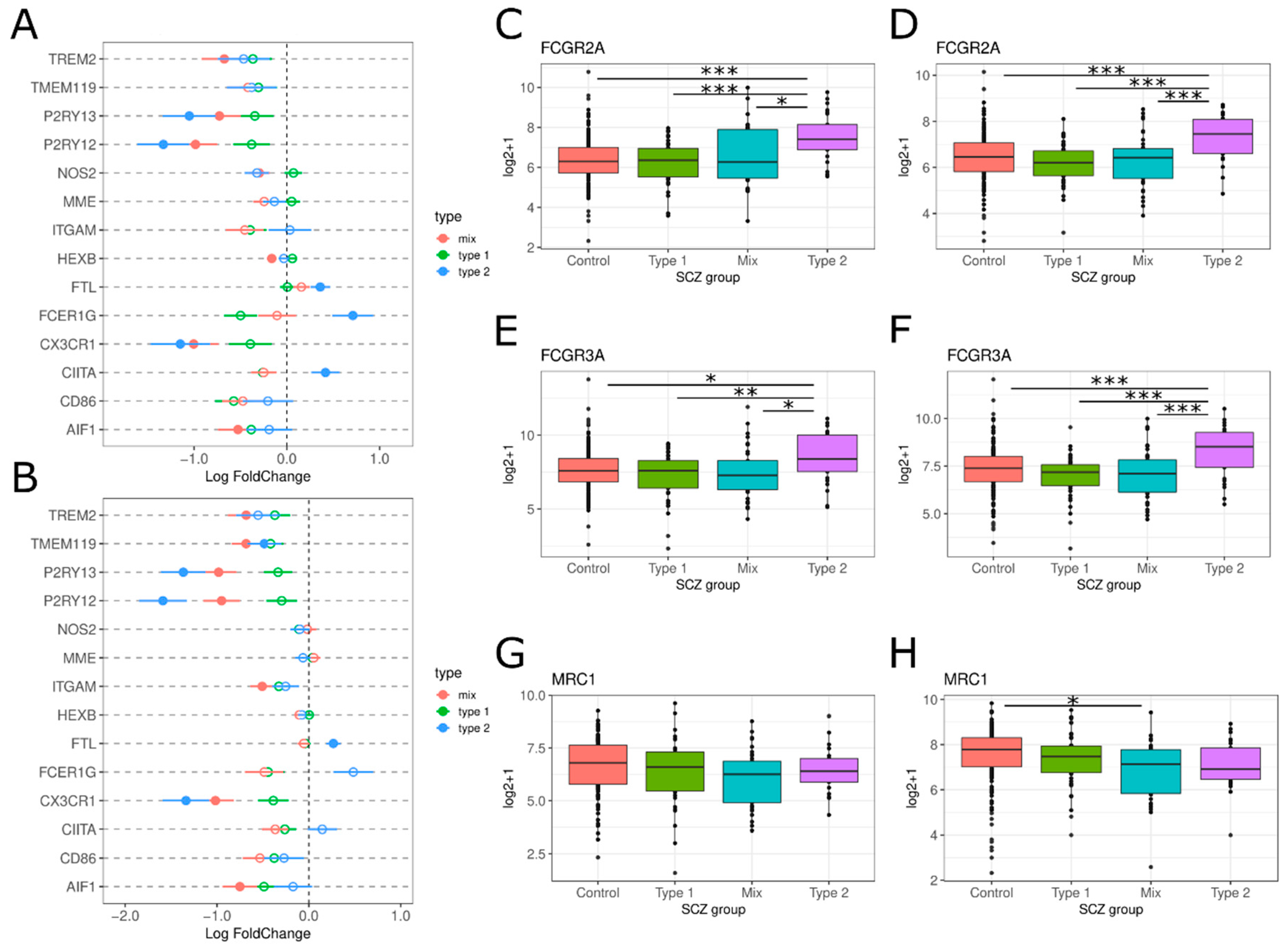
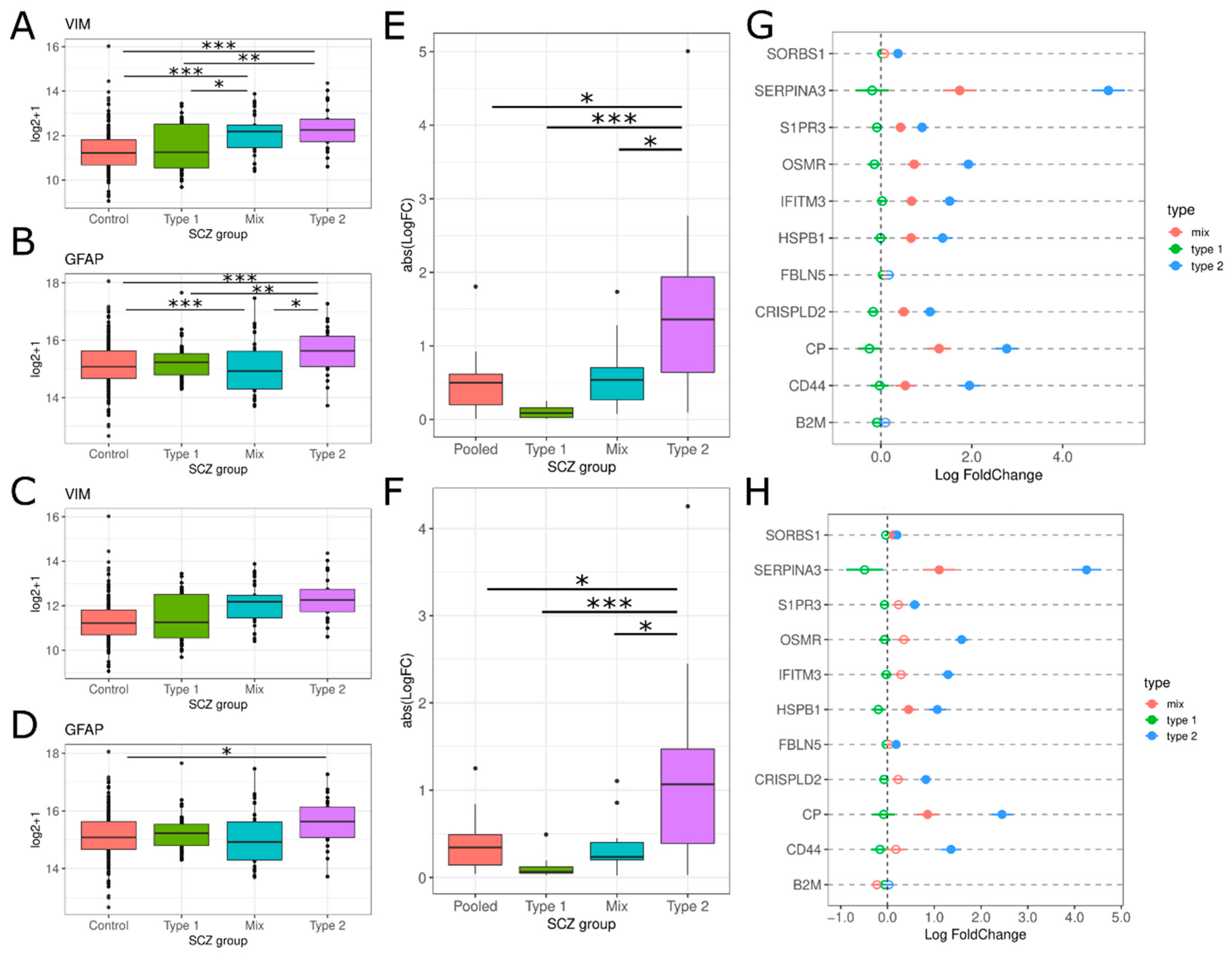
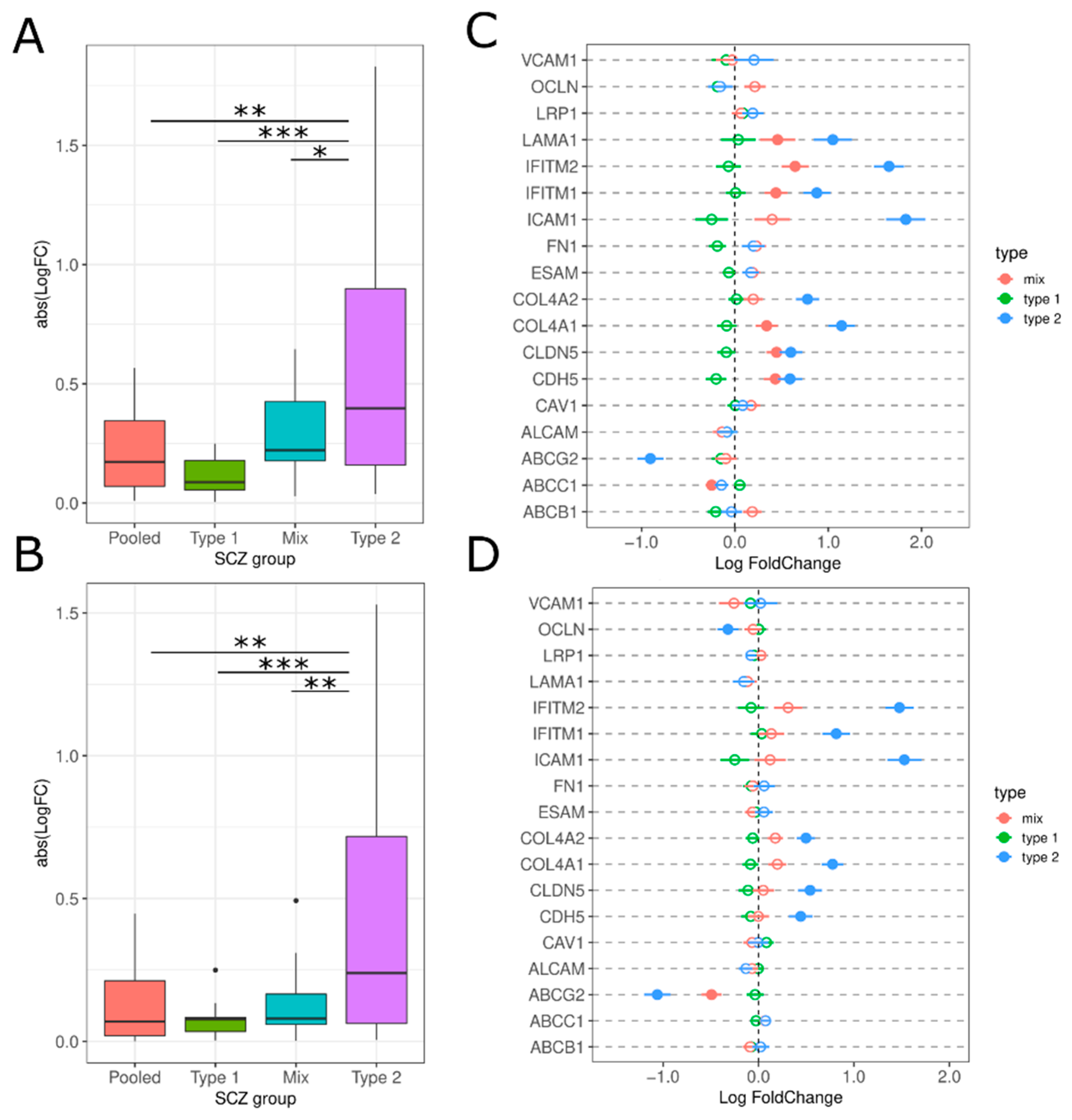
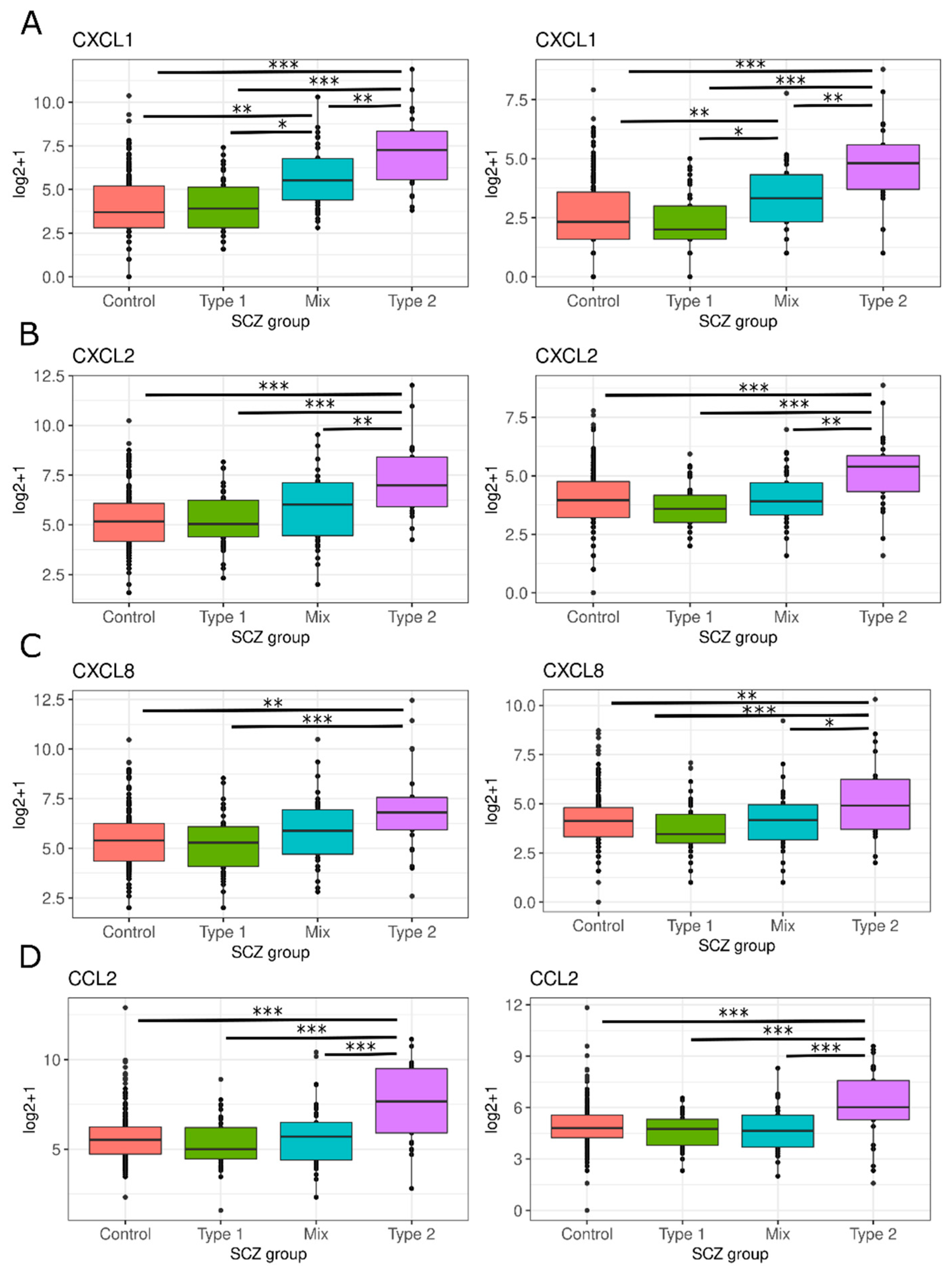
3.2. Neuroinflammation-Related Gene Expression in Type 1 and Type 2 Schizophrenics
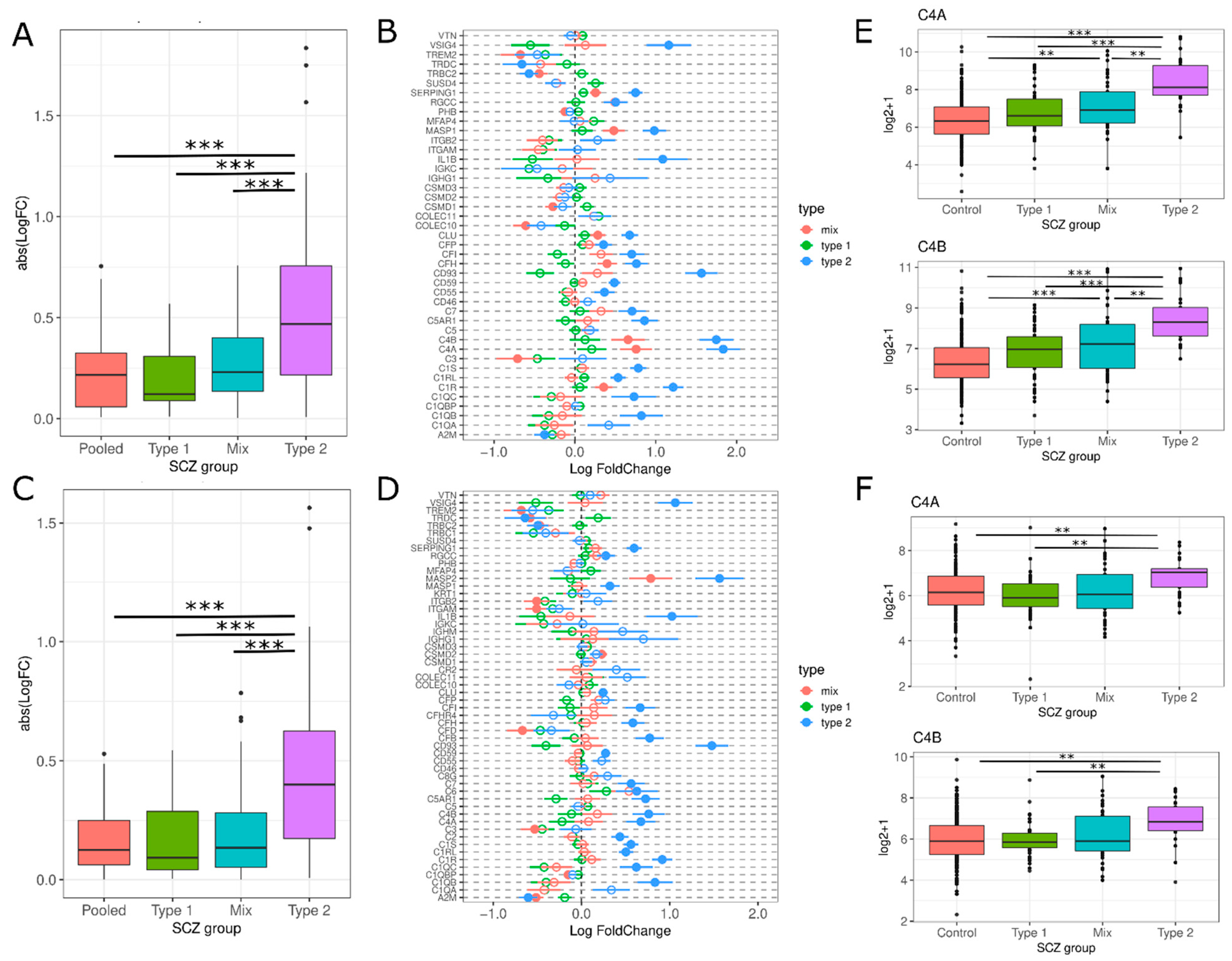
3.2.1. Activation of Complement Cascade System
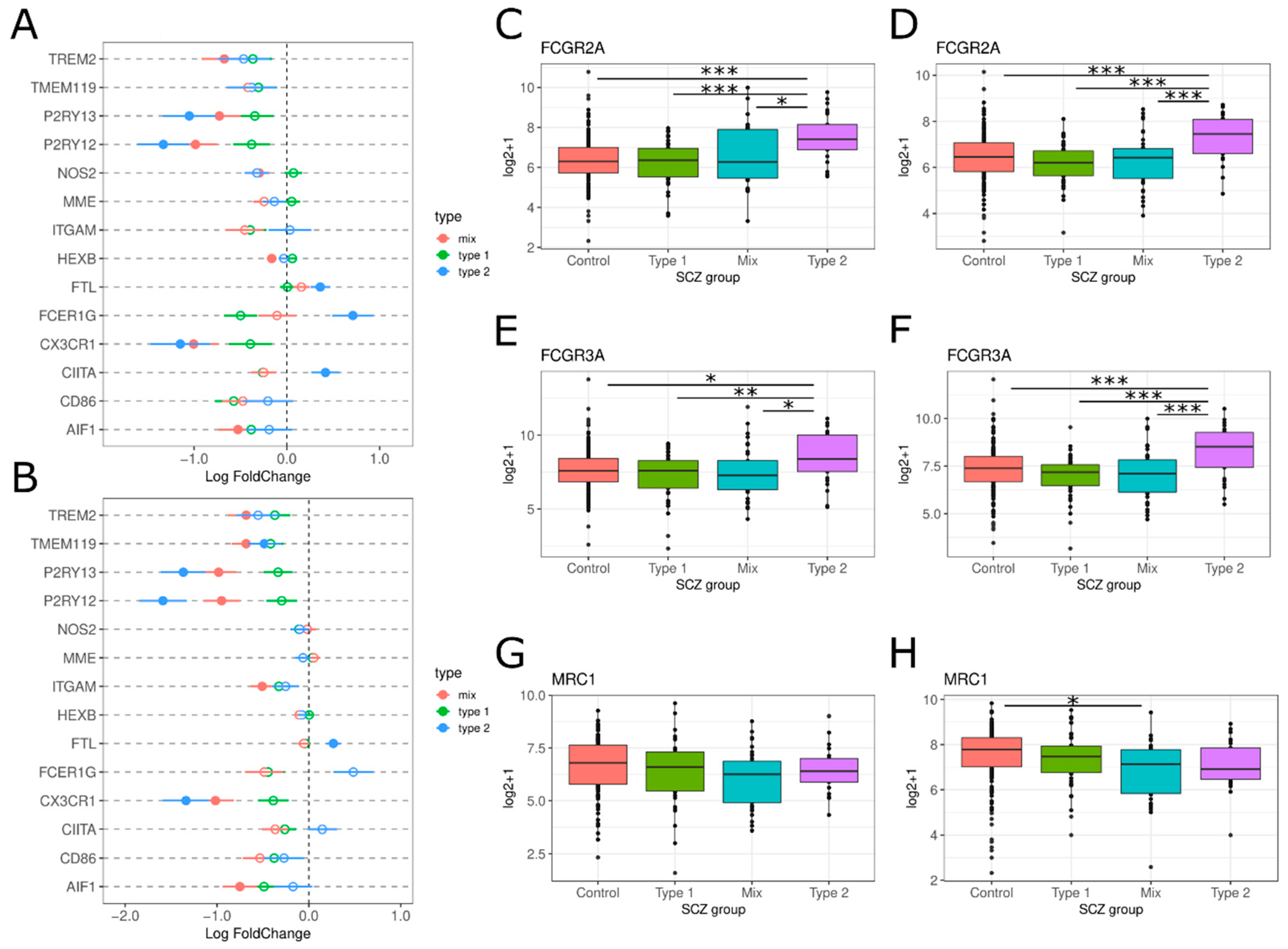
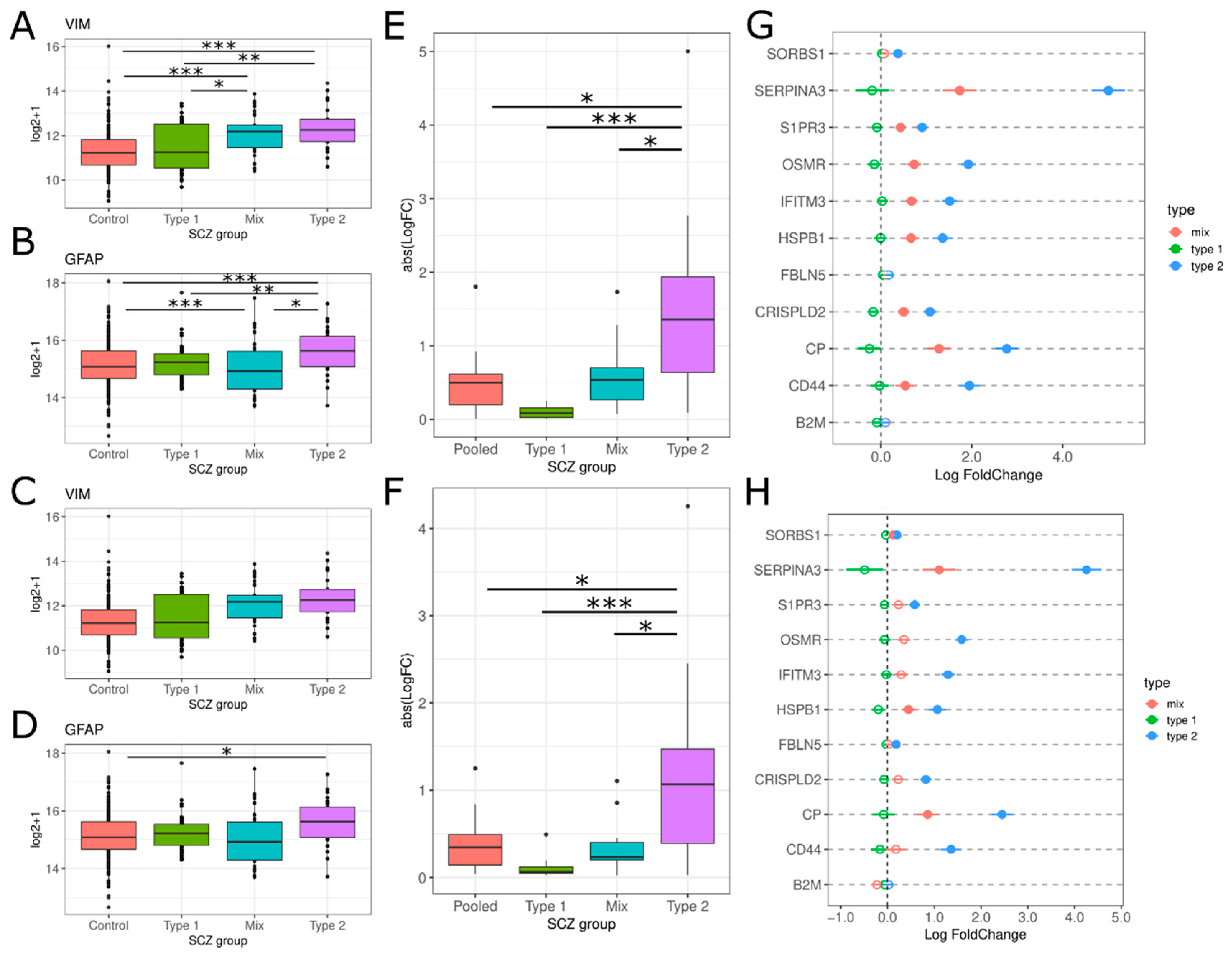
3.2.2. Glial Cell Activation
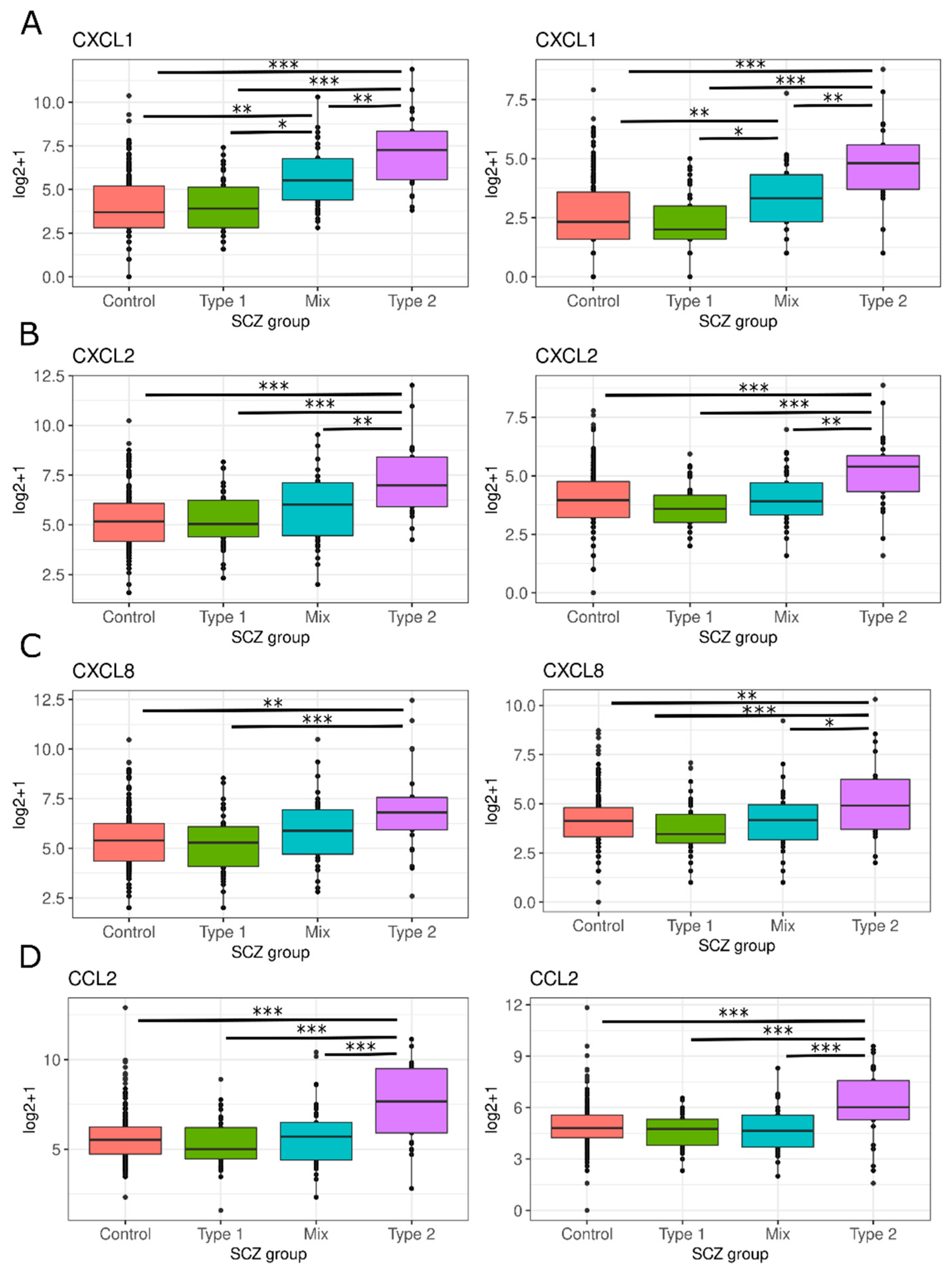
3.2.3. Secretion of Pro-Inflammatory Mediators
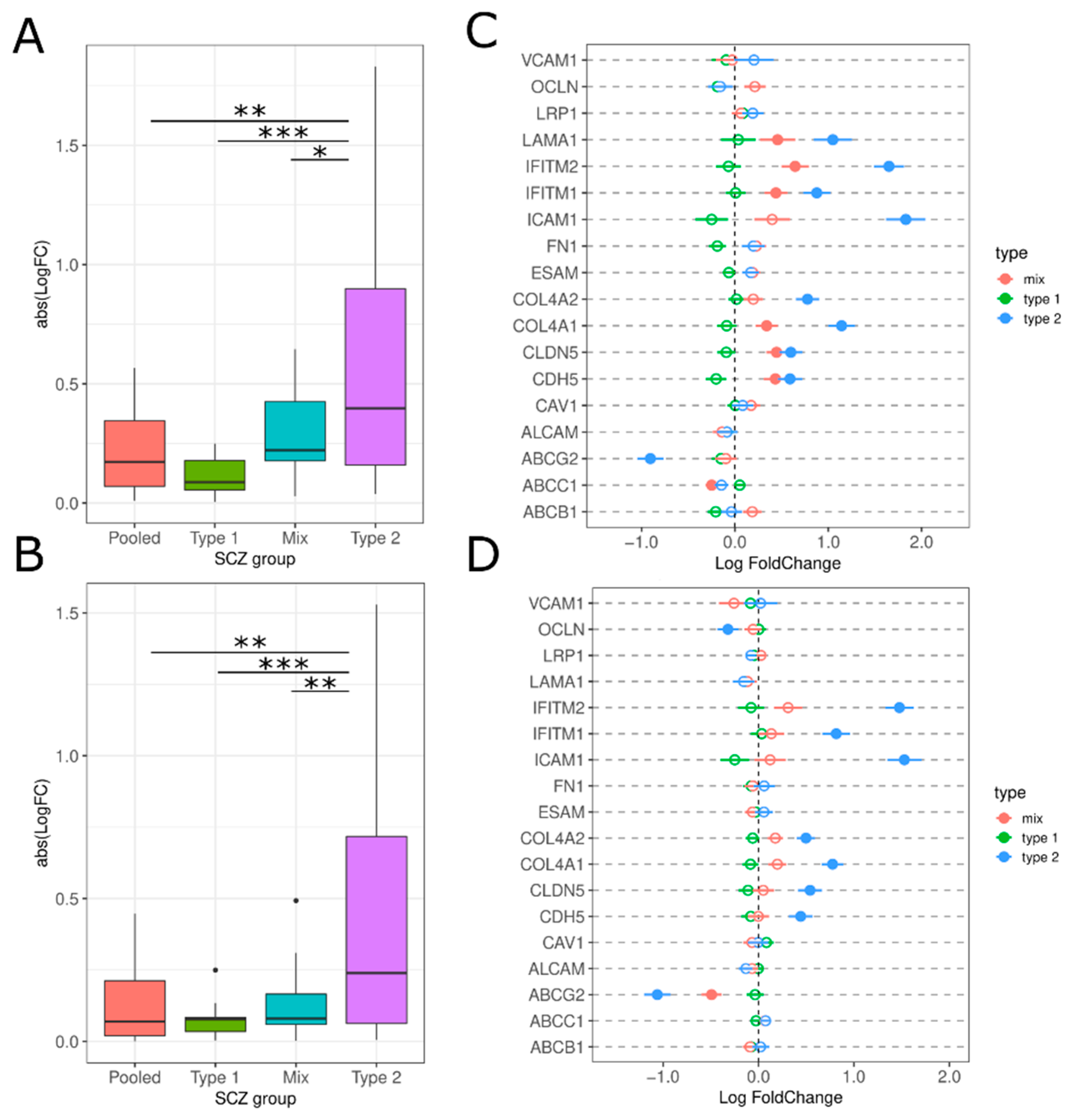
3.2.4. Brain Microvascular Endothelial Cells (BMEC) and BBB Breakdown
3.2.5. Chemokine and Leukocyte Chemotaxis
3.2.6. Peripheral Immune Cell Infiltration
4. Discussion
4.1. Activation of Complement Cascade System
4.2. Glial Cell Activation
4.3. Secretion of Pro-Inflammatory Mediators
4.4. Brain Microvascular Endothelial Cells (BMEC)
4.5. Chemokine and Leukocyte Chemotaxis
4.6. Peripheral Immune Cell Infiltration
4.7. Limitations of This Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirkpatrick, B.; Buchanan, R.W.; Ross, D.E.; Carpenter, J. A separate disease within the syndrome of schizophrenia. Arch. Gen. Psychiatry 2001, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, W.; Kirkpatrick, B. The Heterogeneity of the Long-Term Course of Schizophrenia. Schizophr. Bull. 1988, 14, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Chand, G.B.; Singhal, P.; Dwyer, D.B.; Wen, J.; Erus, G.; Doshi, J.; Srinivasan, D.; Mamourian, E.; Varol, E.; Hwang, G.; et al. Two schizophrenia imaging signatures and and their associations with cognition, psychopathology, and genetics in the general population. medRxiv 2022. [Google Scholar] [CrossRef]
- Xiao, Y.; Liao, W.; Long, Z.; Tao, B.; Zhao, Q.; Luo, C.; Tamminga, C.A.; Keshavan, M.S.; Pearlson, G.D.; Clementz, B.A.; et al. Subtyping Schizophrenia Patients Based on Patterns of Structural Brain Alterations. Schizophr. Bull. 2022, 48, 241–250. [Google Scholar] [CrossRef]
- Liang, S.; Greenwood, T. The Impact of Clincial Heterogeneity in Schizophrenia on Genomic Analyses. Schizophr. Res. 2015, 161, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, E.F.W.; Burgess, J.L.; Granger, R.; Kleinman, J.E.; Rhodes, C.H. DLPFC transcriptome de fi nes two molecular subtypes of schizophrenia. Transl. Psychiatry 2019, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Carlström, E.L.; Niazi, A.; Etemadikhah, M.; Halvardson, J.; Enroth, S.; Stockmeier, C.A.; Rajkowska, G.; Nilsson, B.; Feuk, L. Transcriptome analysis of post-mortem brain tissue reveals up-regulation of the complement cascade in a subgroup of schizophrenia patients. Genes 2021, 12, 1242. [Google Scholar] [CrossRef]
- Takahashi, S. Heterogeneity of schizophrenia: Genetic and symptomatic factors. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 648–652. [Google Scholar] [CrossRef] [Green Version]
- Fillman, S.G.; Cloonan, N.; Catts, V.S.; Miller, L.C.; Wong, J.; Mccrossin, T.; Cairns, M.; Weickert, C.S. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2013, 18, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Fillman, S.G.; Sinclair, D.; Fung, S.J.; Webster, M.J.; Shannon Weickert, C. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl. Psychiatry 2014, 4, e365. [Google Scholar] [CrossRef] [Green Version]
- North, H.F.; Weissleder, C.; Fullerton, J.M.; Sager, R.; Webster, M.J.; Weickert, C.S. A schizophrenia subgroup with elevated inflammation displays reduced microglia, increased peripheral immune cell and altered neurogenesis marker gene expression in the subependymal zone. Transl. Psychiatry 2021, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Wong, J.; Fillman, S.G.; Fung, S.J.; Shannon Weickert, C. Increased expression of astrocyte markers in schizophrenia: Association with neuroinflammation. Aust. N. Z. J. Psychiatry 2014, 48, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.G.; Gonçalves, V.F.; Spain, S.L.; Finucane, H.K.; Raychaudhuri, S.; Kennedy, J.L.; Knight, J. Genome-wide association studies suggest limited immune gene enrichment in schizophrenia compared to 5 autoimmune diseases. Schizophr. Bull. 2016, 42, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Burke, E.E.; Peterson, A.; Shin, J.H.; Straub, R.E.; Rajpurohit, A.; Semick, S.A.; Ulrich, W.S.; Price, A.J.; Valencia, C.; et al. Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 103, ISBN 0000011126. [Google Scholar]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2015, 19, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Voineskos, A.N.; Foussias, G.; Lerch, J.; Felsky, D.; Remington, G.; Rajji, T.K.; Lobaugh, N.; Pollock, B.G.; Mulsant, B.H. Neuroimaging evidence for the deficit subtype of schizophrenia. JAMA Psychiatry 2013, 70, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Koutsouleris, N.; Meisenzahl, E.; Davatzikos, C. Heterogeneity of structural brain changes in subtypes of schizophrenia revealed using magnetic resonance imaging pattern analysis. Schizophr. Bull. 2015, 41, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Nenadic, I.; Yotter, R.A.; Sauer, H.; Gaser, C. Patterns of cortical thinning in different subgroups of schizophrenia. Br. J. Psychiatry 2015, 206, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Chand, G.B.; Dwyer, D.B.; Erus, G.; Sotiras, A.; Varol, E.; Srinivasan, D.; Doshi, J.; Pomponio, R.; Pigoni, A.; Dazzan, P.; et al. Two distinct neuroanatomical subtypes of schizophrenia revealed using machine learning. Brain 2020, 143, 1027–1038. [Google Scholar] [CrossRef]
- Comer, A.L.; Carrier, M.; Tremblay, M.È.; Cruz-Martín, A. The Inflamed Brain in Schizophrenia: The Convergence of Genetic and Environmental Risk Factors That Lead to Uncontrolled Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 274. [Google Scholar] [CrossRef]
- Bishop, J.R.; Zhang, L.; Lizano, P. Inflammation Subtypes and Translating Inflammation-Related Genetic Findings in Schizophrenia and Related Psychoses: A Perspective on Pathways for Treatment Stratification and Novel Therapies. Harv. Rev. Psychiatry 2022, 30, 59–70. [Google Scholar] [CrossRef]
- Gatta, E.; Saudagar, V.; Drnevich, J.; Forrest, M.P.; Auta, J.; Clark, L.V.; Sershen, H.; Smith, R.C.; Grayson, D.R.; Davis, J.M.; et al. Concordance of Immune-Related Markers in Lymphocytes and Prefrontal Cortex in Schizophrenia. Schizophr. Bull. Open 2021, 2, sgab002. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kesteren, C.F.M.G.; Gremmels, H.; De Witte, L.D.; Hol, E.M.; Van Gool, A.R.; Falkai, P.G.; Kahn, R.S.; Sommer, I.E.C. Immune involvement in the pathogenesis of schizophrenia: A meta-analysis on postmortem brain studies. Transl. Psychiatry 2017, 7, e1075-11. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Pi, X.N.; Hu, N.; Wang, X.; Xiao, Y.; Li, S.; Sweeney, J.A.; Bishop, J.R.; Gong, Q.; Xie, D.; et al. Subtypes of schizophrenia identified by multi-omic measures associated with dysregulated immune function. Mol. Psychiatry 2021, 26, 6926–6936. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.Q.; Catts, V.S.; Webster, M.J.; Galletly, C.; Liu, D.; O’Donnell, M.; Weickert, T.W.; Weickert, C.S. Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol. Psychiatry 2020, 25, 761–775. [Google Scholar] [CrossRef] [Green Version]
- Lipska, B.K.; Deep-Soboslay, A.; Weickert, C.S.; Hyde, T.M.; Martin, C.E.; Herman, M.M.; Kleinman, J.E. Critical Factors in Gene Expression in Postmortem Human Brain: Focus on Studies in Schizophrenia. Biol. Psychiatry 2006, 60, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.E.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol. Psychiatry 2018, 23, 1496–1505. [Google Scholar] [CrossRef]
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Woo, J.J.; Pouget, J.G.; Zai, C.C.; Kennedy, J.L. The complement system in schizophrenia: Where are we now and what’s next? Mol. Psychiatry 2020, 25, 114–130. [Google Scholar] [CrossRef]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef] [Green Version]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediators Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef]
- Pong, S.; Karmacharya, R.; Sofman, M.; Bishop, J.R.; Lizano, P. The Role of Brain Microvascular Endothelial Cell and Blood-Brain Barrier Dysfunction in Schizophrenia. Complex Psychiatry 2020, 6, 30–46. [Google Scholar] [CrossRef]
- Le Thuc, O.; Blondeau, N.; Nahon, J.L.; Rovère, C. The complex contribution of chemokines to neuroinflammation: Switching from beneficial to detrimental effects. Ann. N. Y. Acad. Sci. 2015, 1351, 127–140. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shahir, M.; Taheri, M.; Salimi, A. A review on the role of chemokines in the pathogenesis of systemic lupus erythematosus. Cytokine 2021, 146, 375–406. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2016, 20, 136–144. [Google Scholar] [CrossRef]
- Merikangas, A.K.; Shelly, M.; Knighton, A.; Kotler, N.; Tanenbaum, N.; Almasy, L. What genes are differentially expressed in individuals with schizophrenia? A systematic review. Mol. Psychiatry 2022, 27, 1373–1383. [Google Scholar] [CrossRef]
- Arloth, J.; Bader, D.M.; Röh, S.; Altmann, A. Re-Annotator: Annotation pipeline for microarray probe sequences. PLoS ONE 2015, 10, e0139516. [Google Scholar] [CrossRef]
- Rey, R.; Suaud-Chagny, M.F.; Bohec, A.L.; Dorey, J.M.; d’Amato, T.; Tamouza, R.; Leboyer, M. Overexpression of complement component C4 in the dorsolateral prefrontal cortex, parietal cortex, superior temporal gyrus and associative striatum of patients with schizophrenia. Brain. Behav. Immun. 2020, 90, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comer, A.L.; Jinadasa, T.; Sriram, B.; Phadke, R.A.; Kretsge, L.N.; Nguyen, T.P.H.; Antognetti, G.; Gilbert, J.P.; Lee, J.; Newmark, E.R.; et al. Increased Expression of Schizophrenia-Associated Gene C4 Leads to Hypoconnectivity of Prefrontal Cortex and Reduced Social Interaction. PLoS Biol. 2020, 18, e3000604. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Yalcin, E.; Presumey, J.; Aw, E.; Ma, M.; Whelan, C.W.; Stevens, B.; McCarroll, S.A.; Carroll, M.C. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 2021, 24, 214–224. [Google Scholar] [CrossRef]
- Snijders, G.J.L.J.; van Zuiden, W.; Sneeboer, M.A.M.; Berdenis van Berlekom, A.; van der Geest, A.T.; Schnieder, T.; MacIntyre, D.J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. A loss of mature microglial markers without immune activation in schizophrenia. Glia 2021, 69, 1251–1267. [Google Scholar] [CrossRef]
- Bergon, A.; Belzeaux, R.; Comte, M.; Pelletier, F.; Hervé, M.; Gardiner, E.J.; Beveridge, N.J.; Liu, B.; Carr, V.; Scott, R.J.; et al. CX3CR1 is dysregulated in blood and brain from schizophrenia patients. Schizophr. Res. 2015, 168, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Krügel, U.; Kittner, H.; Franke, H.; Illes, P. Stimulation of P2 receptors in the ventral tegmental area enhances dopaminergic mechanisms in vivo. Neuropharmacology 2001, 40, 1084–1093. [Google Scholar] [CrossRef]
- Trendelenburg, A.U.; Bultman, R. P2 Receptor Mediated Inhibition of Dopamine Release in Rat Neostriatum. Neuroscience 2000, 96, 249–252. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Yamashita, H.; Ohshita, T.; Sawamoto, N.; Nakamura, S. ATP increases extracellular dopamine level through stimulation of P2Y purinoceptors in the rat striatum. Brain Res. 1995, 691, 205–212. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain. Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Saetre, P.; Emilsson, L.; Axelsson, E.; Kreuger, J.; Lindholm, E.; Jazin, E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 2007, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Arion, D.; Unger, T.; Lewis, D.A.; Levitt, P.; Mirnics, K. Molecular Evidence for Increased Expression of Genes Related to Immune and Chaperone Function in the Prefrontal Cortex in Schizophrenia. Biol. Psychiatry 2007, 62, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Corsi-Zuelli, F.; Deakin, B. Impaired regulatory T cell control of astroglial overdrive and microglial pruning in schizophrenia. Neurosci. Biobehav. Rev. 2021, 125, 637–653. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Miller, B.J. Inflammation and schizophrenia. Schizophr. Bull. 2013, 39, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Momtazmanesh, S.; Zare-Shahabadi, A.; Rezaei, N. Cytokine Alterations in Schizophrenia: An Updated Review. Front. Psychiatry 2019, 10, 892. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; Maclean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Trojanowski, J.Q.; Gur, R.E.; Blackwell, P.; Han, L.Y.; Choi, C. Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch. Gen. Psychiatry 1998, 55, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.; Jaffe, A.E.; Chen, Q.; Shin, J.H.; Kleinman, J.E.; Hyde, T.M.; Weinberger, D.R. Investigating the neuroimmunogenic architecture of schizophrenia. Mol. Psychiatry 2018, 23, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. The role of intercellular adhesion molecule-1 in the pathogenesis of psychiatric disorders. Front. Pharmacol. 2019, 10, 1251. [Google Scholar] [CrossRef] [PubMed]
- Poller, B.; Drewe, J.; Krähenbühl, S.; Huwyler, J.; Gutmann, H. Regulation of BCRP (ABCG2) and P-Glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell. Mol. Neurobiol. 2010, 30, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Katsel, P.; Roussos, P.; Pletnikov, M.; Haroutunian, V. Microvascular anomaly conditions in psychiatric disease. Schizophrenia angiogenesis connection. Neurosci. Biobehav. Rev. 2017, 77, 327–339. [Google Scholar] [CrossRef]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 2016, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Nishiura, K.; Ichikawa-Tomikawa, N.; Sugimoto, K.; Kunii, Y.; Kashiwagi, K.; Tanaka, M.; Yokoyama, Y.; Hino, M.; Sugino, T.; Yabe, H.; et al. PKA activation and endothelial claudin-5 breakdown in the schizophrenic prefrontal cortex. Oncotarget 2017, 8, 93382–93391. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Volk, D.W.; Chitrapu, A.; Edelson, J.R.; Roman, K.M.; Moroco, A.E.; Lewis, D.A. Molecular Mechanisms and Timing of Cortical Immune Activation in Schizophrenia. Am. J. Psychiatry 2015, 172, 1112–1121. [Google Scholar] [CrossRef]
- Pierson, E.R.; Wagner, C.A.; Goverman, J.M. The contribution of neutrophils to CNS autoimmunity. Clin. Immunol. 2018, 189, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Kim, Y.M.; Lee, J.; Park, J.; Lee, J.E.; Hyun, Y.M. Neutrophils Return to Bloodstream Through the Brain Blood Vessel After Crosstalk With Microglia During LPS-Induced Neuroinflammation. Front. Cell Dev. Biol. 2020, 8, 613733. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Coe, C.L.; Birdsill, A.C.; Bendlin, B.B.; Colman, R.J.; Alexander, A.L.; Allison, D.B.; Weindruch, R.H.; Johnson, S.C. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. Age 2013, 35, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, D.; Zhang, P.; Wu, G.; Cao, L.; Shen, Y. Serum Levels in Neuroleptic-Free Schizophrenia: Association with Psychopathology. Schizophr. Res. 2002, 57, 247–258. [Google Scholar] [CrossRef]
- Janssen, K.; Rickert, M.; Clarner, T.; Beyer, C.; Kipp, M. Absence of CCL2 and CCL3 Ameliorates Central Nervous System Grey Matter But Not White Matter Demyelination in the Presence of an Intact Blood–Brain Barrier. Mol. Neurobiol. 2016, 53, 1551–1564. [Google Scholar] [CrossRef]
- Hong, S.; Lee, E.E.; Martin, A.S.; Soontornniyomkij, B.; Soontornniyomkij, V.; Achim, C.L.; Reuter, C.; Irwin, M.R.; Eyler, L.T.; Jeste, D.V. Abnormalities in chemokine levels in schizophrenia and their clinical correlates. Schizophr. Res. 2017, 181, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Beumer, W.; Drexhage, R.C.; De Wit, H.; Versnel, M.A.; Drexhage, H.A.; Cohen, D. Increased level of serum cytokines, chemokines and adipokines in patients with schizophrenia is associated with disease and metabolic syndrome. Psychoneuroendocrinology 2012, 37, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, D.H.; Lee, S.; Yantis, J.; Valdez, C.; Paredes, R.M.; Braida, N.; Velligan, D.; Walss-Bass, C. Differential correlations between inflammatory cytokines and psychopathology in veterans with schizophrenia: Potential role for IL-17 pathway. Schizophr. Res. 2013, 151, 29–35. [Google Scholar] [CrossRef]
- Reale, M.; Patruno, A.; De Lutiis, M.A.; Pesce, M.; Felaco, M.; Di Giannantonio, M.; Di Nicola, M.; Grilli, A. Dysregulation of chemo-cytokine production in schizophrenic patients versus healthy controls. BMC Neurosci. 2011, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Zakharyan, R.; Boyajyan, A.; Arakelyan, A.; Melkumova, M.; Mrazek, F.; Petrek, M. Monocyte chemoattractant protein-1 in schizophrenia: -2518A/G genetic variant and protein levels in Armenian population. Cytokine 2012, 58, 351–354. [Google Scholar] [CrossRef]
- Asevedo, E.; Gadelha, A.; Noto, C.; Mansur, R.B.; Zugman, A.; Belangero, S.I.N.; Berberian, A.A.; Scarpato, B.S.; Leclerc, E.; Teixeira, A.Ô.L.; et al. Impact of peripheral levels of chemokines, BDNF and oxidative markers on cognition in individuals with schizophrenia. J. Psychiatr. Res. 2013, 47, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Bellani, M.; Isola, M.; Bergami, A.; Marinelli, V.; Dusi, N.; Rambaldelli, G.; Tansella, M.; Maria Finardi, A.; Martino, G.; et al. Increased M1/decreased M2 signature and signs of Th1/Th2 shift in chronic patients with bipolar disorder, but not in those with schizophrenia. Transl. Psychiatry 2014, 4, e406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicola, M.; Cattaneo, A.; Hepgul, N.; Di Forti, M.; Aitchison, K.J.; Janiri, L.; Murray, R.M.; Dazzan, P.; Pariante, C.M.; Mondelli, V. Serum and gene expression profile of cytokines in first-episode psychosis. Brain. Behav. Immun. 2013, 31, 90–95. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Reis, H.J.; Nicolato, R.; Brito-Melo, G.; Correa, H.; Teixeira, M.M.; Romano-Silva, M.A. Increased serum levels of CCL11/eotaxin in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Rummel, C.; Inoue, W.; Poole, S.; Luheshi, G.N. Leptin regulates leukocyte recruitment into the brain following systemic lps-induced inflammation. Mol. Psychiatry 2010, 15, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Aguliar-Valles, A.; Kim, J.; Jung, S.; Woodside, B.; Luheshi, G.N. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol. Psychiatry 2014, 19, 599–606. [Google Scholar] [CrossRef]
- Núñez, C.; Stephan-Otto, C.; Usall, J.; Bioque, M.; Lobo, A.; González-Pinto, A.; Pina-Camacho, L.; Vieta, E.; Castro-Fornieles, J.; Rodriguez-Jimenez, R.; et al. Neutrophil count is associated with reduced gray matter and enlarged ventricles in first-episode psychosis. Schizophr. Bull. 2019, 45, 846–858. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Childers, E.; Bowen, E.F.W.; Rhodes, C.H.; Granger, R. Immune-Related Genomic Schizophrenic Subtyping Identified in DLPFC Transcriptome. Genes 2022, 13, 1200. https://doi.org/10.3390/genes13071200
Childers E, Bowen EFW, Rhodes CH, Granger R. Immune-Related Genomic Schizophrenic Subtyping Identified in DLPFC Transcriptome. Genes. 2022; 13(7):1200. https://doi.org/10.3390/genes13071200
Chicago/Turabian StyleChilders, Eva, Elijah F. W. Bowen, C. Harker Rhodes, and Richard Granger. 2022. "Immune-Related Genomic Schizophrenic Subtyping Identified in DLPFC Transcriptome" Genes 13, no. 7: 1200. https://doi.org/10.3390/genes13071200
APA StyleChilders, E., Bowen, E. F. W., Rhodes, C. H., & Granger, R. (2022). Immune-Related Genomic Schizophrenic Subtyping Identified in DLPFC Transcriptome. Genes, 13(7), 1200. https://doi.org/10.3390/genes13071200