
Integrated Analyses of DNA Methylation and Gene Expression of Rainbow Trout Muscle under Variable Ploidy and Muscle Atrophy Conditions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Population and Muscle Sampling
2.2. RNA Sequencing and Analyses
2.3. Methyl-MiniSeq Sequencing and Analyses
3. Results and Discussion
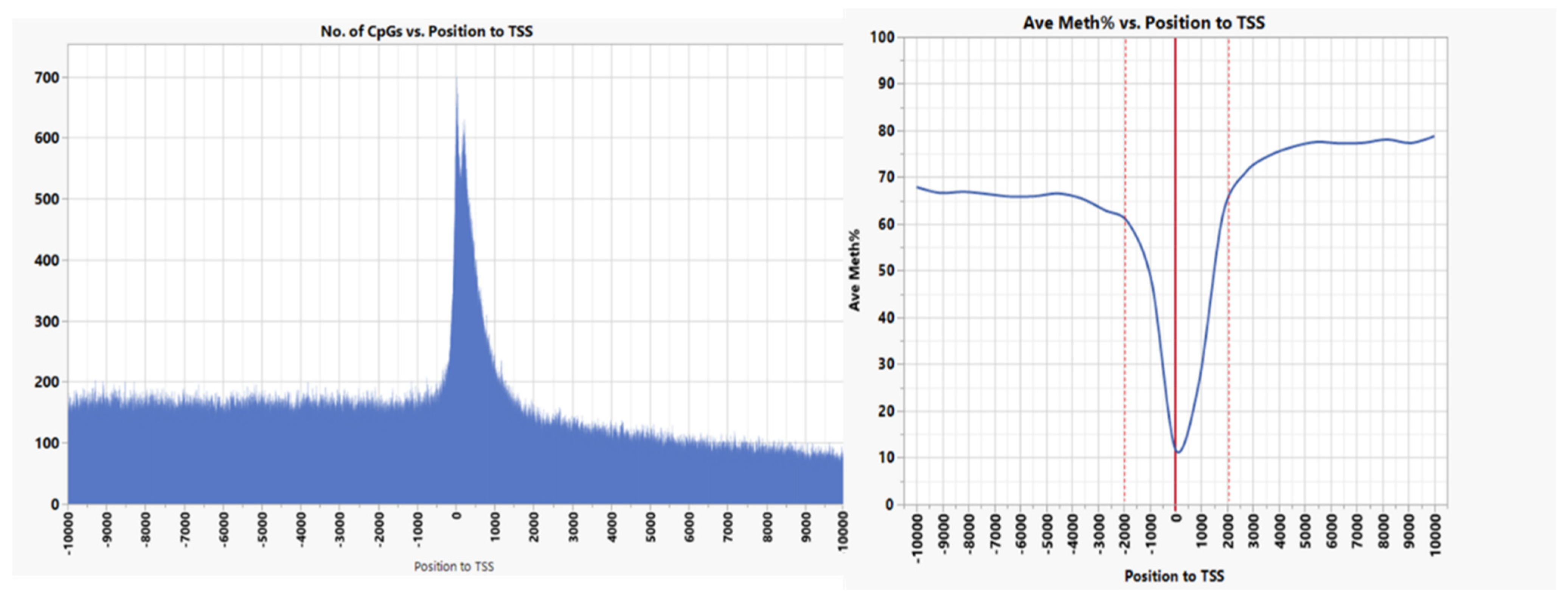
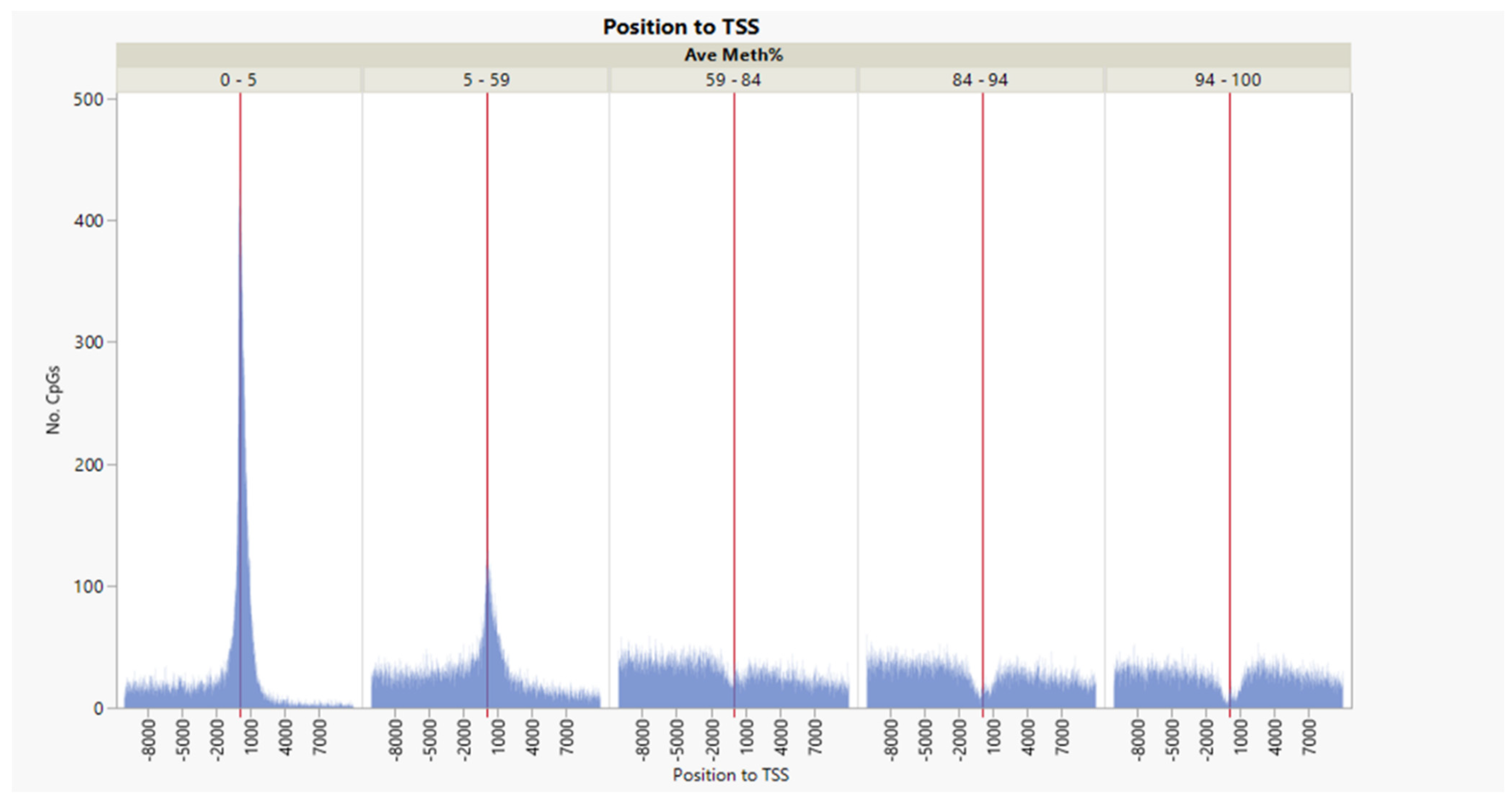
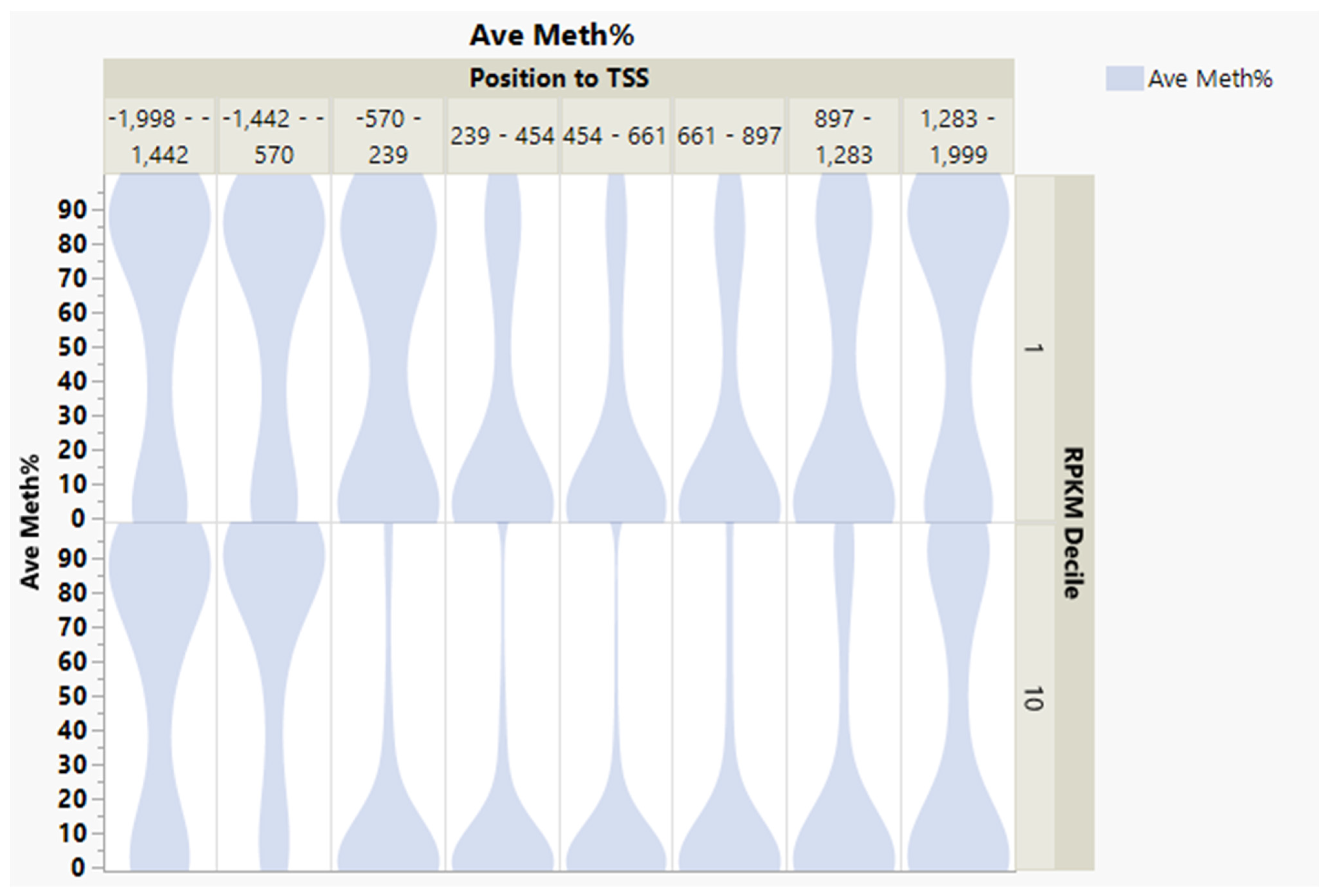
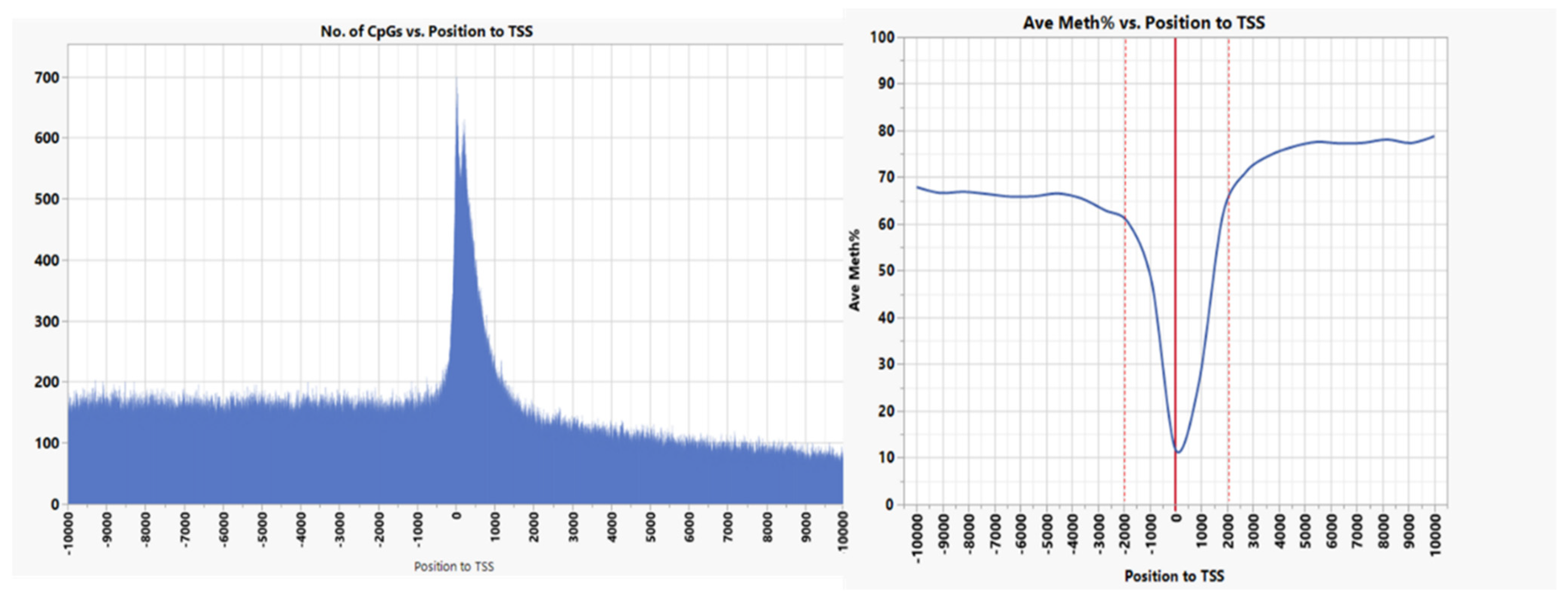
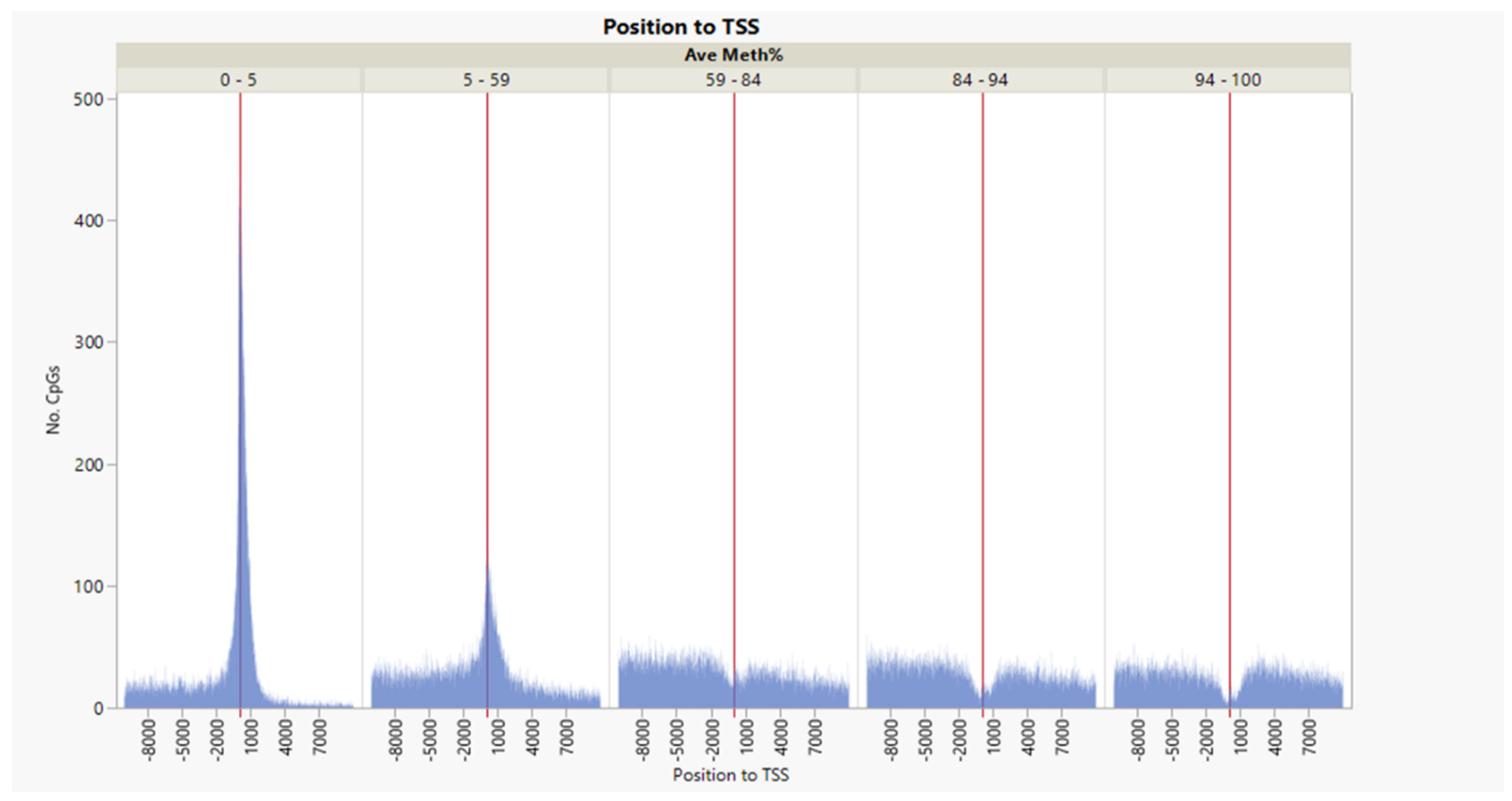
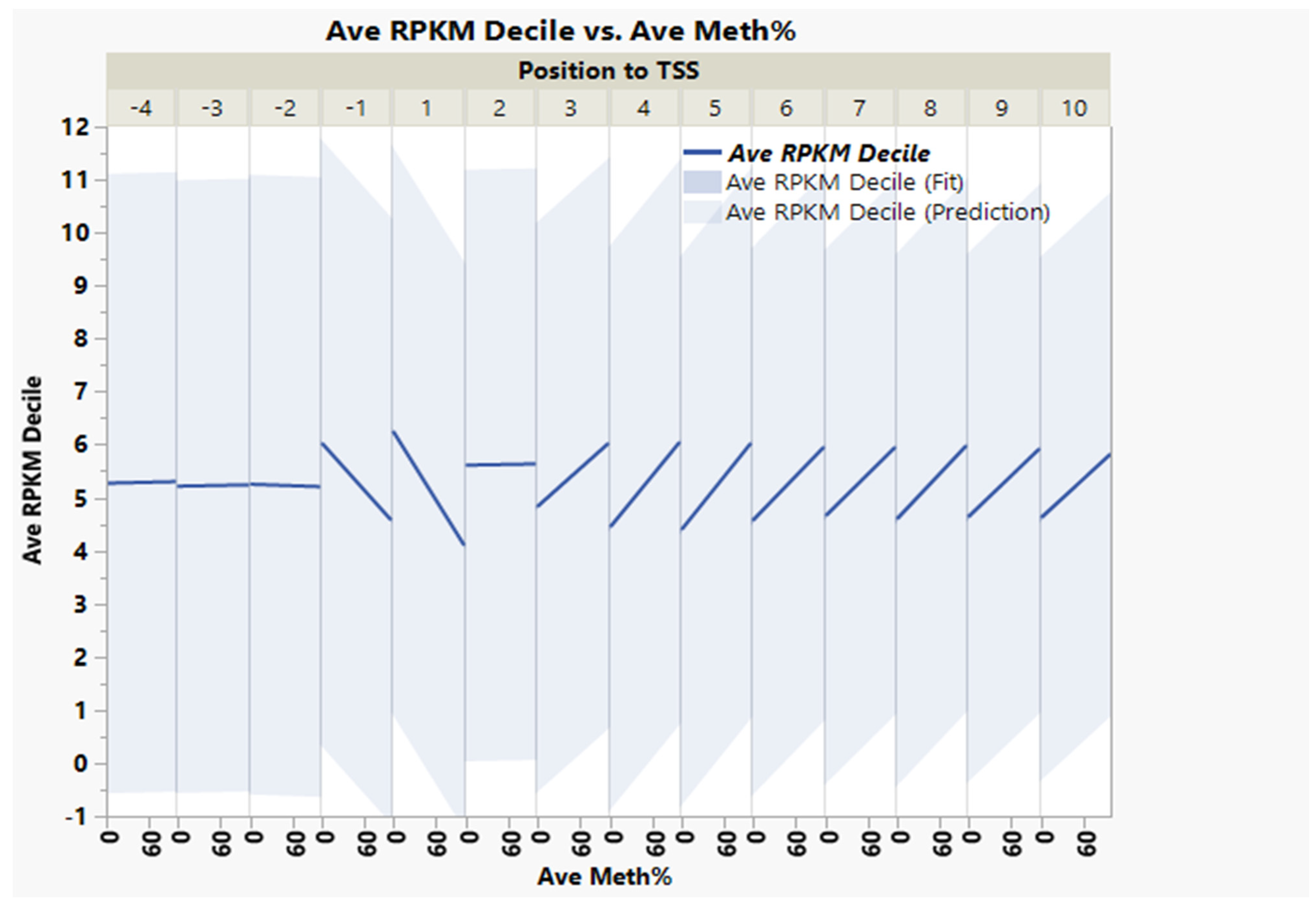
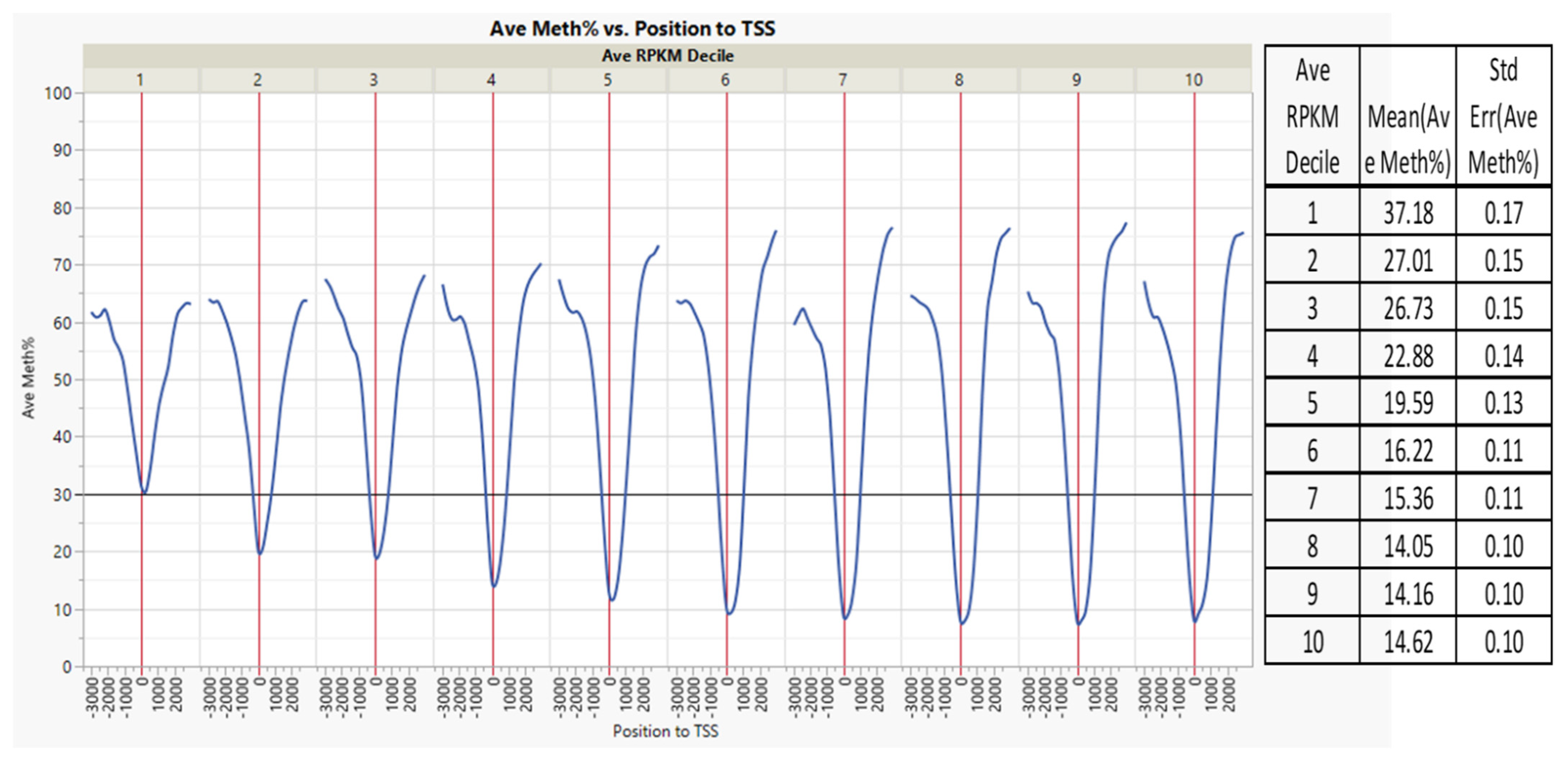
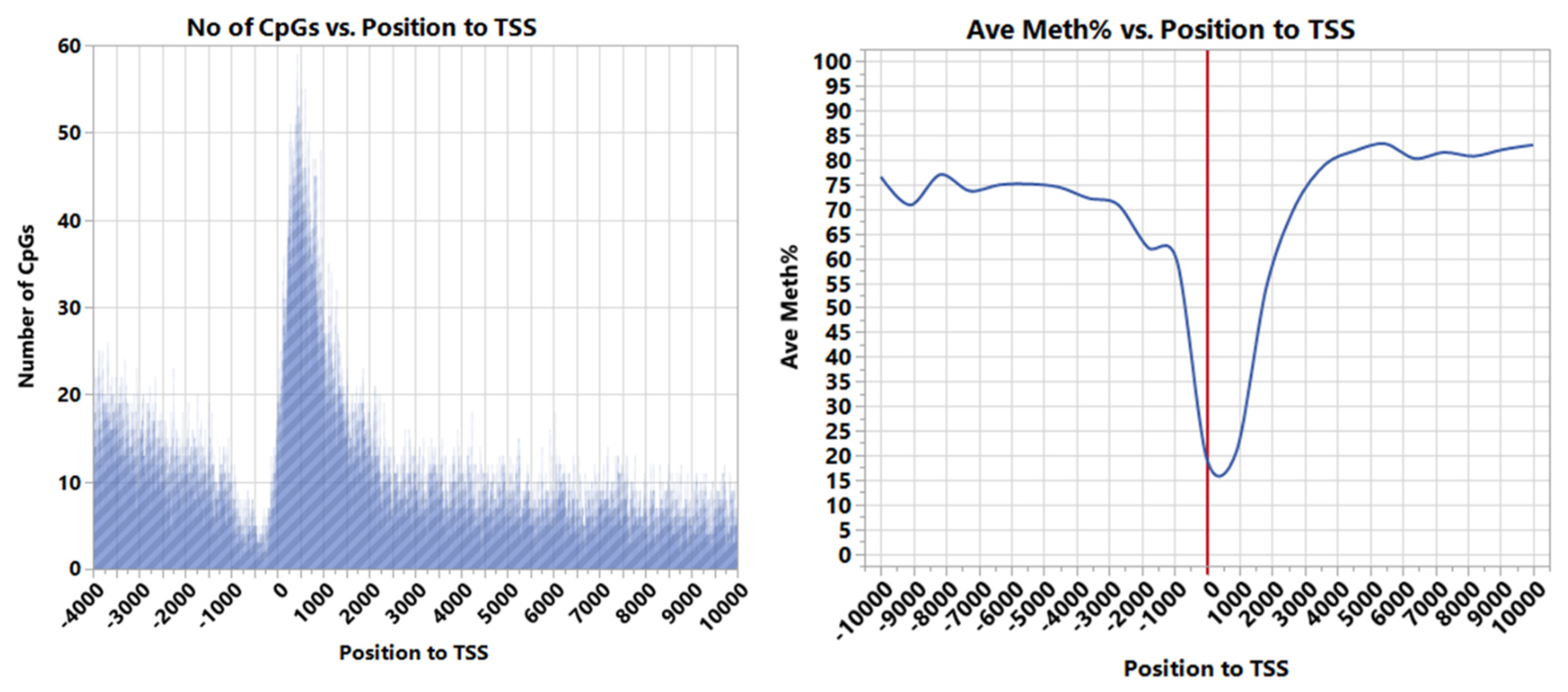
3.1. CpG Dinucleotide Content in the Rainbow Trout Genome
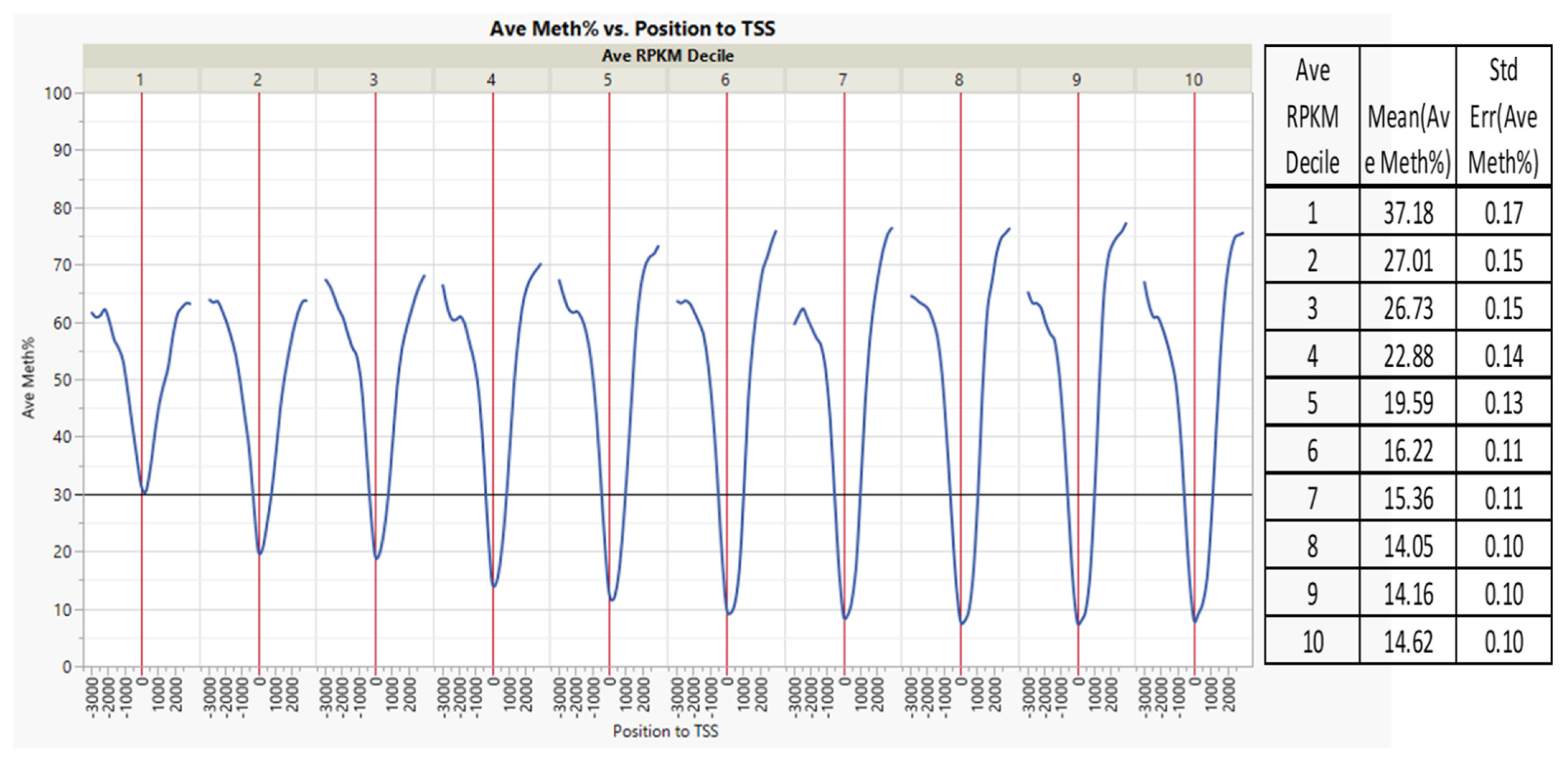
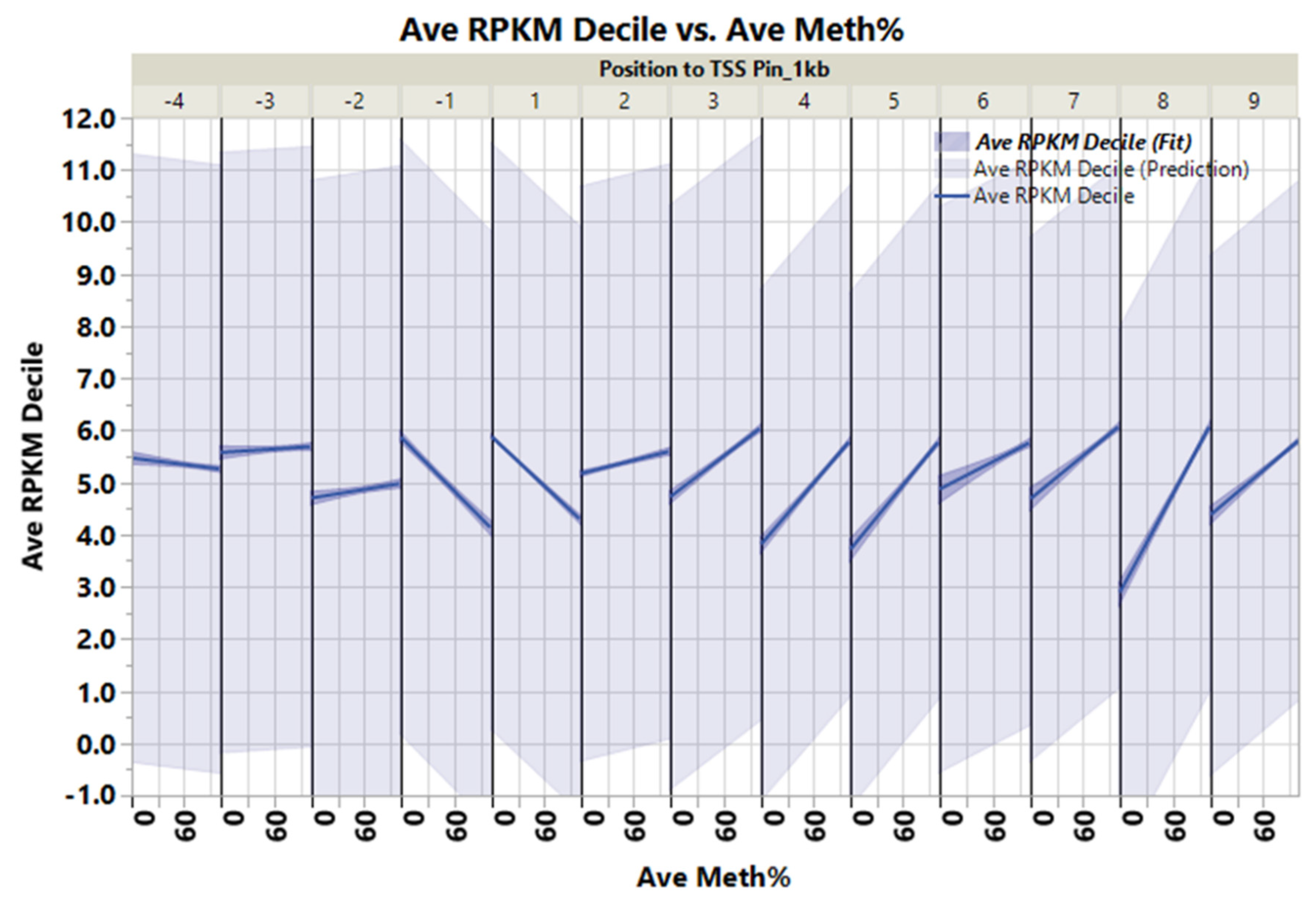
3.2. Non-Island Genic CpGs and Their Associated Gene Expression
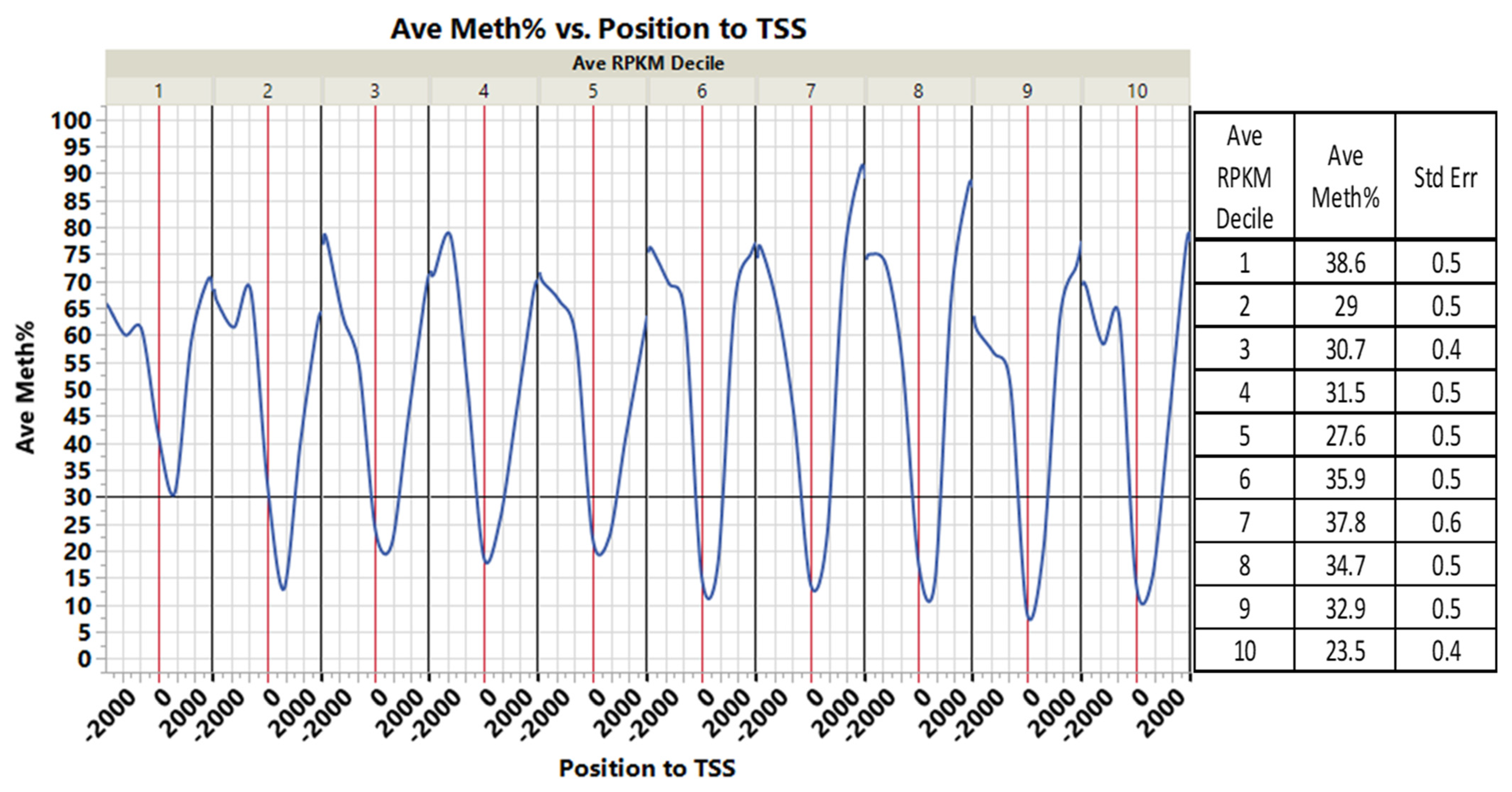
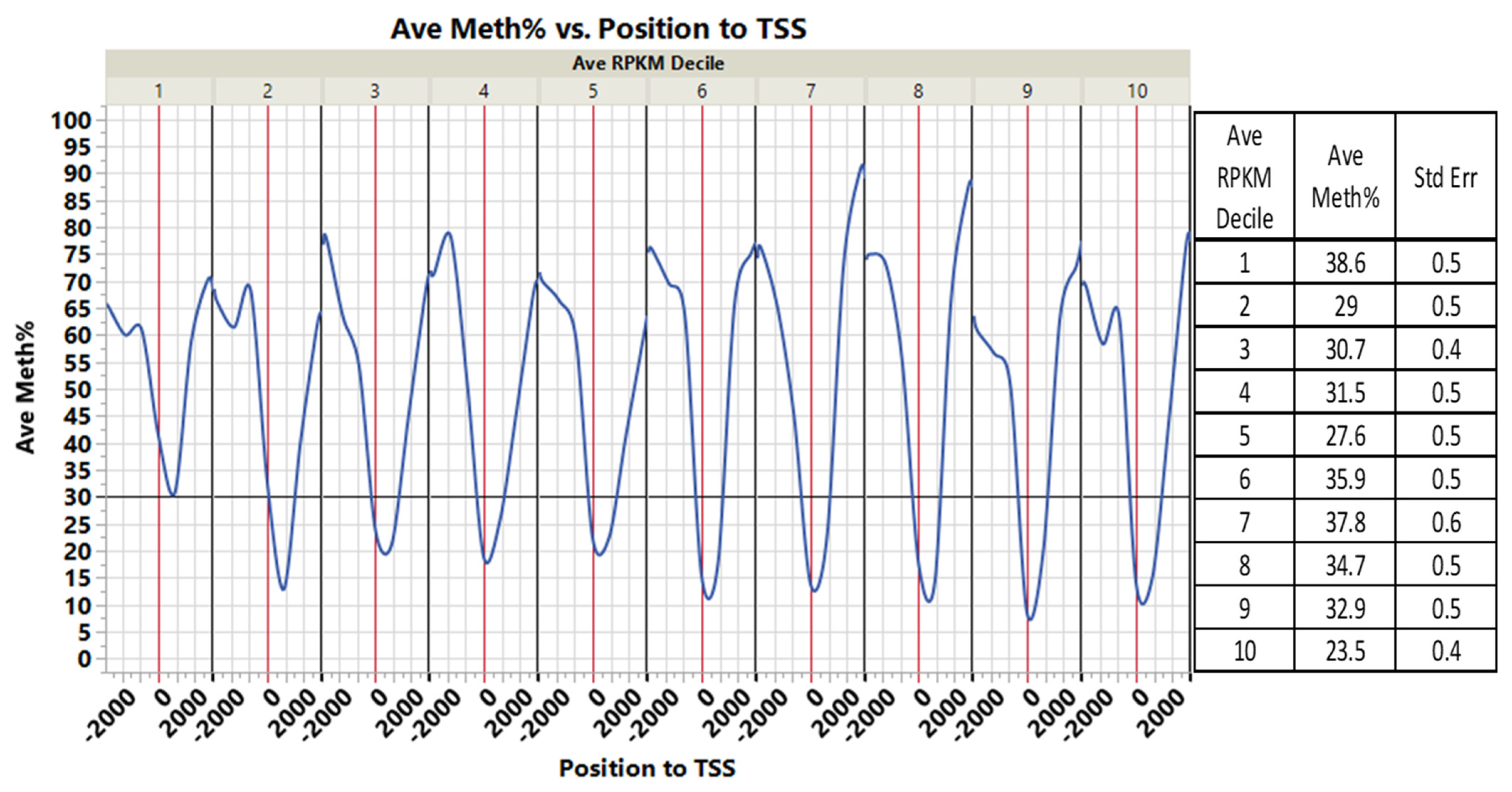
3.3. CpGs Islands within or near Genes and Their Associated Gene Expression
3.4. Differential DNA Methylation and Gene Expression between Diploid/Gravid and Triploid/Sterile Fish
3.4.1. Differentially Methylated (DM) CpGs
3.4.2. Integrated Analysis of Differential Gene Expression and DNA Methylation
3.4.3. Apoptosis and Epigenetic Regulation Genes
3.4.4. Autophagy, Glycolysis, Collagen Metabolism, Cell Membrane Functions Genes
3.4.5. Homeobox Proteins
3.4.6. Other Epigenetic Markers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Availability of Data and Materials
Consent for Publication
References
- Aquaculture Genomics, G.; Breeding, W.; Abdelrahman, H.; ElHady, M.; Alcivar-Warren, A.; Allen, S.; Al-Tobasei, R.; Bao, L.; Beck, B.; Blackburn, H.; et al. Aquaculture genomics, genetics and breeding in the United States: Current status, challenges, and priorities for future research. BMC Genom. 2017, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Magadan, S.; Waldbieser, G.C.; Youngblood, R.C.; Wheeler, P.A.; Scheffler, B.E.; Thorgaard, G.H.; Palti, Y. A long reads-based de-novo assembly of the genome of the Arlee homozygous line reveals chromosomal rearrangements in rainbow trout. G3 Bethesda 2021, 11, jkab052. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Thorgaard, G.H.; Salem, M. PacBio Iso-Seq Improves the Rainbow Trout Genome Annotation and Identifies Alternative Splicing Associated with Economically Important Phenotypes. Front. Genet. 2021, 12, 683408. [Google Scholar] [CrossRef] [PubMed]
- Lallias, D.; Bernard, M.; Ciobotaru, C.; Dechamp, N.; Labbe, L.; Goardon, L.; Le Calvez, J.M.; Bideau, M.; Fricot, A.; Prezelin, A.; et al. Sources of variation of DNA methylation in rainbow trout: Combined effects of temperature and genetic background. Epigenetics 2021, 16, 1031–1052. [Google Scholar] [CrossRef] [PubMed]
- Gavery, M.R.; Nichols, K.M.; Berejikian, B.A.; Tatara, C.P.; Goetz, G.W.; Dickey, J.T.; Van Doornik, D.M.; Swanson, P. Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment. Genes 2019, 10, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhuang, Z.X.; Sun, Y.Q.; Chen, Q.; Zheng, X.Y.; Liang, Y.T.; Mahboob, S.; Wang, Q.; Zhang, R.; Al-Ghanim, K.A.; et al. Changes in DNA methylation during epigenetic-associated sex reversal under low temperature in Takifugu rubripes. PLoS ONE 2019, 14, e0221641. [Google Scholar] [CrossRef]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inf. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Rishi, V.; Bhattacharya, P.; Chatterjee, R.; Rozenberg, J.; Zhao, J.; Glass, K.; Fitzgerald, P.; Vinson, C. CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20311–20316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, P.P.; Wang, J.; Cleveland, B.; Yao, J. 17beta-Estradiol Increases Non-CpG Methylation in Exon 1 of the Rainbow Trout (Oncorhynchus mykiss) MyoD Gene. Mar. Biotechnol. 2017, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Mori, M.; Inagawa, M.; Miyata, K.; Hashimoto, N.; Tanaka, S.; Asahara, H. Dnmt3a Regulates Proliferation of Muscle Satellite Cells via p57Kip2. PLoS Genet. 2016, 12, e1006167. [Google Scholar] [CrossRef] [Green Version]
- Tajrishi, M.M.; Shin, J.; Hetman, M.; Kumar, A. DNA methyltransferase 3a and mitogen-activated protein kinase signaling regulate the expression of fibroblast growth factor-inducible 14 (Fn14) during denervation-induced skeletal muscle atrophy. J. Biol. Chem. 2014, 289, 19985–19999. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.A.; Cervenka, I.; Khatri, B.; Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Perry, R.A., Jr.; Brown, L.A.; Haynie, W.S.; Wiggs, M.P.; et al. Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol. Genom. 2018, 50, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Zykovich, A.; Hubbard, A.; Flynn, J.M.; Tarnopolsky, M.; Fraga, M.F.; Kerksick, C.; Ogborn, D.; MacNeil, L.; Mooney, S.D.; Melov, S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Matzke, M.A.; Mittelsten Scheid, O.; Matzke, A.J. Rapid structural and epigenetic changes in polyploid and aneuploid genomes. Bioessays 1999, 21, 761–767. [Google Scholar] [CrossRef]
- Madlung, A. Polyploidy and its effect on evolutionary success: Old questions revisited with new tools. Heredity 2013, 110, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ali, A.; Hou, F.; Wu, T.; Guo, D.; Zeng, X.; Wang, F.; Zhao, H.; Chen, X.; Xu, P.; et al. Effects of ploidy variation on promoter DNA methylation and gene expression in rice (Oryza sativa L.). BMC Plant Biol. 2018, 18, 314. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, Y.; Qin, H.; Zhao, C.; Yang, L.; Yu, T.; Zhang, Y.; Xu, T.; Qin, Q.; Liu, S. Genetic and Epigenetic Changes Are Rapid Responses of the Genome to the Newly Synthesized Autotetraploid Carassius auratus. Front. Genet. 2020, 11, 576260. [Google Scholar] [CrossRef] [PubMed]
- Covelo-Soto, L.; Leunda, P.M.; Perez-Figueroa, A.; Moran, P. Genome-wide methylation study of diploid and triploid brown trout (Salmo trutta L.). Anim. Genet. 2015, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Kenney, P.B.; Rexroad, C.E., 3rd; Yao, J. Microarray gene expression analysis in atrophying rainbow trout muscle: A unique nonmammalian muscle degradation model. Physiol. Genom. 2006, 28, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.; Kenney, P.B.; Rexroad, C.E.; Yao, J. Molecular characterization of muscle atrophy and proteolysis associated with spawning in rainbow trout. Comp. Biochem. Physiol. Part D Genom. Proteom. 2006, 1, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Kenney, P.B.; Rexroad, C.E., 3rd; Yao, J. Proteomic signature of muscle atrophy in rainbow trout. J. Proteom. 2010, 73, 778–789. [Google Scholar] [CrossRef]
- Paneru, B.; Ali, A.; Al-Tobasei, R.; Kenney, B.; Salem, M. Crosstalk among lncRNAs, microRNAs and mRNAs in the muscle ‘degradome’ of rainbow trout. Sci. Rep. 2018, 8, 8416. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Farre, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Rosello, L.; Alba, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.Y.; Xia, J.H.; Lin, G.; Wang, L.; Lin, V.C.; Yue, G.H. Genome-wide methylation analysis identified sexually dimorphic methylated regions in hybrid tilapia. Sci. Rep. 2016, 6, 35903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Hartono, S.R.; Korf, I.F.; Chedin, F. GC skew is a conserved property of unmethylated CpG island promoters across vertebrates. Nucleic Acids Res. 2015, 43, 9729–9741. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hoshida, Y.; Sadler, K.C. Comparative Epigenomic Profiling of the DNA Methylome in Mouse and Zebrafish Uncovers High Interspecies Divergence. Front. Genet. 2016, 7, 110. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, H.; Schulz, R. Protection of CpG islands against de novo DNA methylation during oogenesis is associated with the recognition site of E2f1 and E2f2. Epigenetics Chromatin 2014, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Baubec, T.; Schubeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, V.N.; Gossmann, T.I.; Schachtschneider, K.M.; Garroway, C.J.; Madsen, O.; Verhoeven, K.J.; de Jager, V.; Megens, H.J.; Warren, W.C.; Minx, P.; et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 2016, 7, 10474. [Google Scholar] [CrossRef] [Green Version]
- Heberle, E.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Beemelmanns, A.; Ribas, L.; Anastasiadi, D.; Moraleda-Prados, J.; Zanuzzo, F.S.; Rise, M.L.; Gamperl, A.K. DNA Methylation Dynamics in Atlantic Salmon (Salmo salar) Challenged With High Temperature and Moderate Hypoxia. Front. Mar. Sci. 2021, 7, 604878. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Diaz, N.; Piferrer, F. Small ocean temperature increases elicit stage-dependent changes in DNA methylation and gene expression in a fish, the European sea bass. Sci. Rep. 2017, 7, 12401. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadi, D.; Vandeputte, M.; Sanchez-Baizan, N.; Allal, F.; Piferrer, F. Dynamic epimarks in sex-related genes predict gonad phenotype in the European sea bass, a fish with mixed genetic and environmental sex determination. Epigenetics 2018, 13, 988–1011. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Han, L.; Su, B.; Li, W.H.; Zhao, Z. Features and trend of loss of promoter-associated CpG islands in the human and mouse genomes. Mol. Biol. Evol. 2007, 24, 1991–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Zhao, Z. Comparative analysis of CpG islands in four fish genomes. Comp. Funct. Genom. 2008, 2008, 565631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.K.; Sims, D.; Heger, A.; Blackledge, N.P.; Kutter, C.; Wright, M.L.; Grutzner, F.; Odom, D.T.; Patient, R.; Ponting, C.P.; et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. Elife 2013, 2, e00348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Zhang, Q.; Deng, X.; Zhang, X.; Chen, C.; Lv, D.; Li, Y.; Li, D.; Zhang, Y.; Li, P.; et al. Conflicts of CpG density and DNA methylation are proximally and distally involved in gene regulation in human and mouse tissues. Epigenetics 2018, 13, 721–741. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.; Taylor, M.S.; Engstrom, P.G.; Frith, M.C.; et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010, 11, R70. [Google Scholar] [CrossRef] [Green Version]
- Yamakoshi, K.; Shimoda, N. De novo DNA methylation at the CpG island of the zebrafish no tail gene. Genesis 2003, 37, 195–202. [Google Scholar] [CrossRef]
- Simo-Mirabet, P.; Perera, E.; Calduch-Giner, J.A.; Perez-Sanchez, J. Local DNA methylation helps to regulate muscle sirtuin 1 gene expression across seasons and advancing age in gilthead sea bream (Sparus aurata). Front. Zool. 2020, 17, 15. [Google Scholar] [CrossRef]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated With Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef]
- Rauch, T.; Wang, Z.; Zhang, X.; Zhong, X.; Wu, X.; Lau, S.K.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc. Natl. Acad. Sci. USA 2007, 104, 5527–5532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Al-Tobasei, R.; Lourenco, D.; Leeds, T.; Kenney, B.; Salem, M. Genome-Wide Association Study Identifies Genomic Loci Affecting Filet Firmness and Protein Content in Rainbow Trout. Front. Genet. 2019, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Lopez, S.; Nagandla, H.; Soibam, B.; Benham, A.; Nguyen, J.; Valenzuela, N.; Wu, H.J.; Burns, A.R.; Rasmussen, T.L.; et al. Mouse myofibers lacking the SMYD1 methyltransferase are susceptible to atrophy, internalization of nuclei and myofibrillar disarray. Dis. Model. Mech. 2016, 9, 347–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene, N. CDK2AP1 Cyclin Dependent Kinase 2 Associated Protein 1 [Homo sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 15 March 2022).
- Zhang, L.; Yang, Y.; Li, B.; Scott, I.C.; Lou, X. The DEAD-box RNA helicase Ddx39ab is essential for myocyte and lens development in zebrafish. Development 2018, 145, dev161018. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Huang, X.; Huang, J.; Zheng, Y.; March, M.E.; Li, J.; Wei, Y. The Role of Autophagy in Skeletal Muscle Diseases. Front. Physiol. 2021, 12, 638983. [Google Scholar] [CrossRef]
- Seiliez, I.; Gabillard, J.C.; Riflade, M.; Sadoul, B.; Dias, K.; Averous, J.; Tesseraud, S.; Skiba, S.; Panserat, S. Amino acids downregulate the expression of several autophagy-related genes in rainbow trout myoblasts. Autophagy 2012, 8, 364–375. [Google Scholar] [CrossRef]
- Cordani, M.; Sanchez-Alvarez, M.; Strippoli, R.; Bazhin, A.V.; Donadelli, M. Sestrins at the Interface of ROS Control and Autophagy Regulation in Health and Disease. Oxid. Med. Cell. Longev. 2019, 2019, 1283075. [Google Scholar] [CrossRef]
- Segales, J.; Perdiguero, E.; Serrano, A.L.; Sousa-Victor, P.; Ortet, L.; Jardi, M.; Budanov, A.V.; Garcia-Prat, L.; Sandri, M.; Thomson, D.M.; et al. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat. Commun. 2020, 11, 189. [Google Scholar] [CrossRef]
- Salem, M.; Manor, M.L.; Aussanasuwannakul, A.; Kenney, P.B.; Weber, G.M.; Yao, J. Effect of sexual maturation on muscle gene expression of rainbow trout: RNA-Seq approach. Physiol. Rep. 2013, 1, e00120. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Joshi, S.; Possemato, R.; Abraham, R.T.; Sabatini, D.M.; White, E. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene 2015, 34, 5662–5676. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of Autophagy by Glycolysis in Cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef]
- Raffaello, A.; Laveder, P.; Romualdi, C.; Bean, C.; Toniolo, L.; Germinario, E.; Megighian, A.; Danieli-Betto, D.; Reggiani, C.; Lanfranchi, G. Denervation in murine fast-twitch muscle: Short-term physiological changes and temporal expression profiling. Physiol. Genom. 2006, 25, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Database, G.H.G. P3H4 Gene—GeneCards|SC65 Protein|SC65 Antibody. 2022. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=P3H4 (accessed on 15 March 2022).
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, Y.; Ohshima, N.; Yoshizawa, I.; Kamei, Y.; Mariggio, S.; Okamoto, K.; Maeda, M.; Nogusa, Y.; Fujioka, Y.; Izumi, T.; et al. A novel glycerophosphodiester phosphodiesterase, GDE5, controls skeletal muscle development via a non-enzymatic mechanism. J. Biol. Chem. 2010, 285, 27652–27663. [Google Scholar] [CrossRef] [Green Version]
- Wakayama, Y.; Hirako, S.; Ohtaki, H.; Arata, S.; Jimi, T.; Honda, K. Histopathological and aquaporin7 mRNA expression analyzes in the skeletal and cardiac muscles of obese db/db mice. J. Vet. Med. Sci. 2021, 83, 1155–1160. [Google Scholar] [CrossRef]
- Aussanasuwannakul, A.; Kenney, P.B.; Weber, G.M.; Yao, J.; Slider, S.D.; Manor, M.L.; Salem, M. Effect of sexual maturation on growth, fillet composition, and texture of female rainbow trout (Oncorhynchus mykiss) on a high nutritional plane. Aquaculture 2011, 317, 79–88. [Google Scholar] [CrossRef]
- Pei, Y.F.; Hu, W.Z.; Yang, X.L.; Wei, X.T.; Feng, G.J.; Zhang, H.; Shen, H.; Tian, Q.; Deng, H.W.; Zhang, L. Two functional variants at 6p21.1 were associated with lean mass. Skelet. Muscle 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Database, G.H.G. ANK1 Gene—GeneCards|ANK1 Protein|ANK1 Antibody. 2022. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ANK1 (accessed on 15 March 2022).
- Houghton, L.; Rosenthal, N. Regulation of a muscle-specific transgene by persistent expression of Hox genes in postnatal murine limb muscle. Dev. Dyn. 1999, 216, 385–397. [Google Scholar] [CrossRef]
- Tsumagari, K.; Baribault, C.; Terragni, J.; Chandra, S.; Renshaw, C.; Sun, Z.; Song, L.; Crawford, G.E.; Pradhan, S.; Lacey, M.; et al. DNA methylation and differentiation: HOX genes in muscle cells. Epigenetics Chromatin 2013, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.C.; Gorski, P.P.; Maasar, M.F.; Seaborne, R.A.; Baumert, P.; Brown, A.D.; Kitchen, M.O.; Erskine, R.M.; Dos-Remedios, I.; Voisin, S.; et al. DNA methylation across the genome in aged human skeletal muscle tissue and muscle-derived cells: The role of HOX genes and physical activity. Sci. Rep. 2020, 10, 15360. [Google Scholar] [CrossRef] [PubMed]
- Garcia de la Serrana, D.; Johnston, I.A. Expression of heat shock protein (Hsp90) paralogues is regulated by amino acids in skeletal muscle of Atlantic salmon. PLoS ONE 2013, 8, e74295. [Google Scholar] [CrossRef] [Green Version]
- Database, G.H.G. CCT5 Gene—GeneCards|TCPE Protein|TCPE Antibody. 2022. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCT5 (accessed on 15 March 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
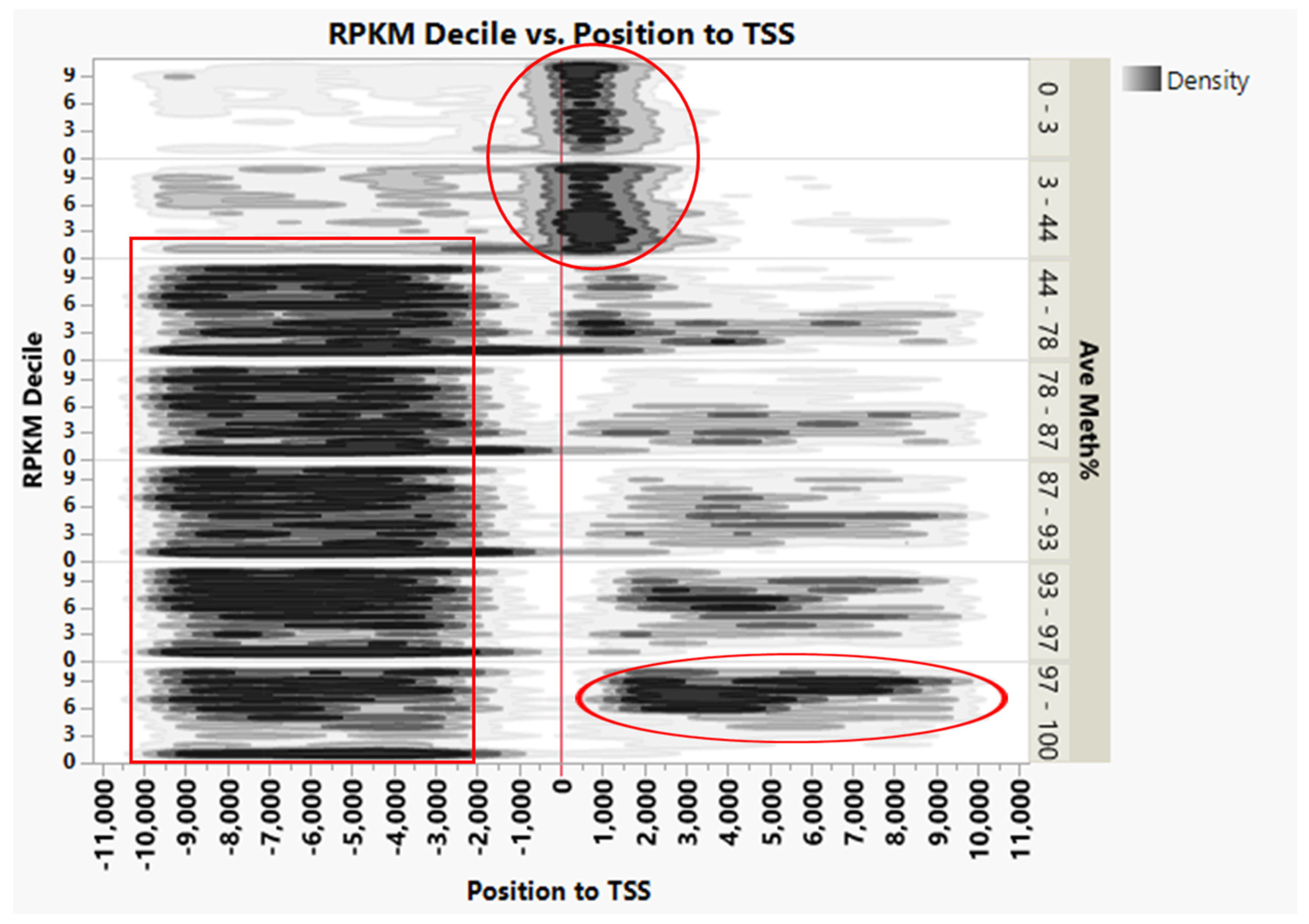{kind=link}
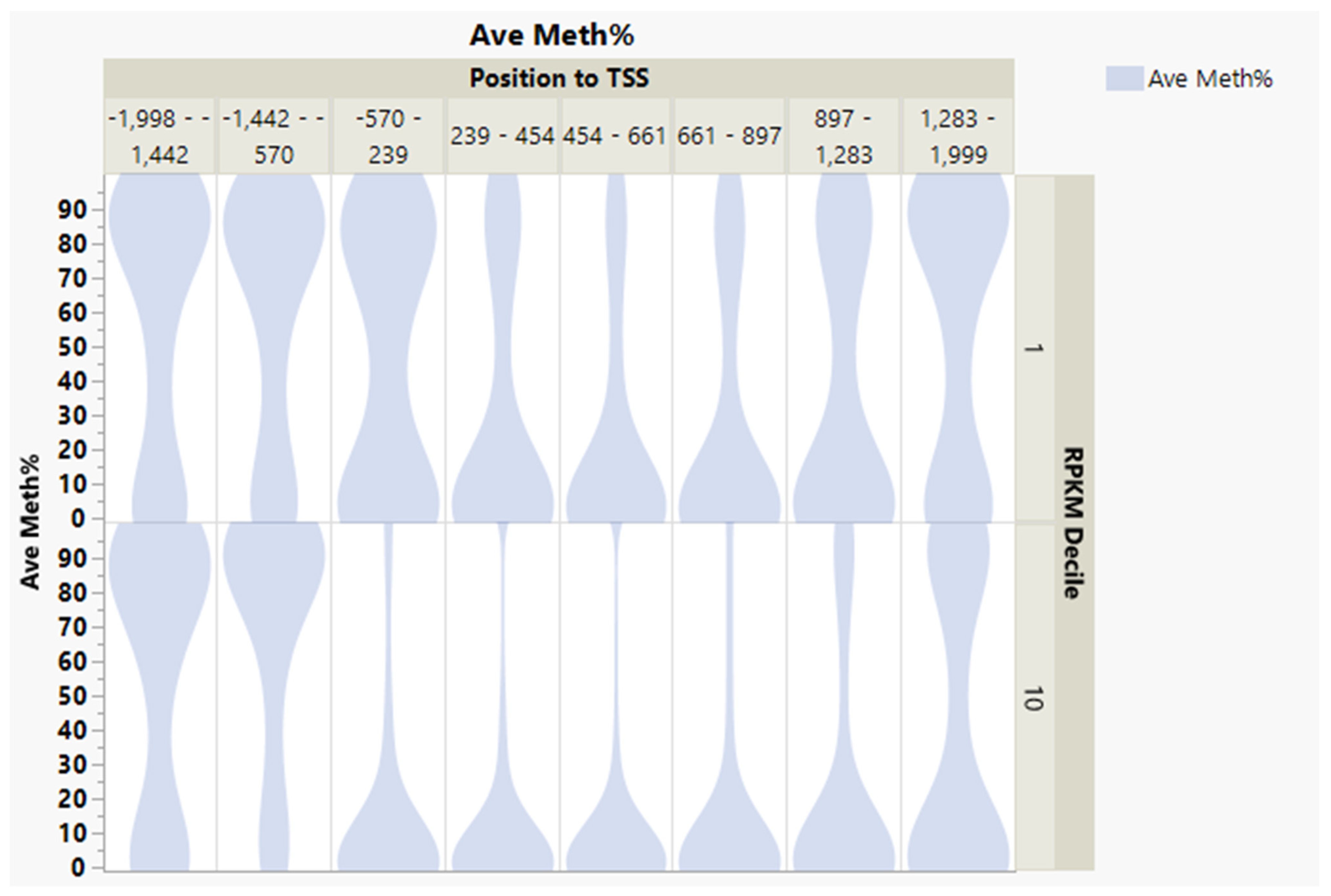{kind=link}
| Source | Term_Name | Term_id | Adj_p Value |
|---|---|---|---|
| GO:MF | DNA-binding transcription factor activity, RNA polymerase II-specific | GO:0000981 | 1.96 × 10−3 |
| GO:MF | Metal ion transmembrane transporter activity | GO:0046873 | 2.24 × 10−2 |
| GO:BP | Homophilic cell adhesion via plasma membrane adhesion molecules | GO:0007156 | 6.60 × 10−5 |
| GO:BP | Cell adhesion | GO:0007155 | 3.18 × 10−2 |
| GO:BP | Biological adhesion | GO:0022610 | 3.18 × 10−2 |
| GO:BP | Cation transport | GO:0006812 | 1.66 × 10−2 |
| GO:BP | Metal ion transport | GO:0030001 | 9.59 × 10−3 |
| GO:BP | Cell-cell adhesion | GO:0098609 | 2.59 × 10−4 |
| GO:BP | Cell–cell adhesion via plasma-membrane adhesion molecules | GO:0098742 | 1.28 × 10−4 |
| Chr Position | Gene/Locus | Position to TSS | Hypo/Hyper Meth (2N–3N) | Meth. Diff p Value | Exp. Fold Change (2N/3N) | Exp. FDR | Exp/Meth R Value | Gene Annotation | TF |
|---|---|---|---|---|---|---|---|---|---|
| Apoptosis and epigenetic regulation | |||||||||
| NC_035082.1_64665435 | LOC110526468/Pdcd5 | −899 | 33.3 | 3.2 × 10−5 | −2.10 | 7.71 × 10−2 | −0.441 | Programmed cell death protein 5 | HES-1 [T01649]; RXR-alpha [T01345]; 3; MYB2 [T02536]; AR [T00040]; USF-1 [T00875]; NF-1 [T00537]; NF-1 [T00539] |
| NC_035082.1_64665694 | LOC110526468/Pdcd6 | −640 | 63.5 | 3.2 × 10−5 | −2.10 | 7.71 × 10−2 | −0.497 | Programmed cell death protein 5 | c-Ets-1 [T00112] |
| NC_035078.1_41106515 | LOC110492018/AEN | 4446 | −48.8 | 7.5 × 10−6 | −3.52 | 1.01 × 10−4 | 0.434 | Apoptosis-enhancing nuclease | E2F-1 [T01542]; C/EBPbeta [T00581]; C/EBPgamma [T00216]; NF-1 [T00535] |
| NC_035081.1_29419783 | LOC110523624/Smyd1 | 3041 | −65.0 | 4.0 × 10−7 | −2.95 | 6.10 × 10−3 | 0.459 | Histone-lysine N-methyl transferase Smyd1 | LyF-1 [T00479]; PTF1-beta [T00701]; myogenin [T00528]; Pax-5 [T00070] |
| NC_035088.1_13620476 | LOC110537077/Cdk2ap1 | −586 | 48.0 | 1.7 × 10−5 | −7.36 | 9.39 × 10−5 | −0.439 | Cyclin-dependent kinase 2-associated protein 1 | USF1 [T00874]; USF1 [T00874]; c-Myc [T00140]; RXR-beta [T01349]; RXR-beta [T01349]; PIF3 [T04492]; PIF3 [T04492]; SREBP-1c [T01562]; CBF1 [T00080]; CBF1 [T00080]; JunD [T00437]; JunD [T00437] |
| NC_035089.1_41352275 | LOC110486510/Ddx39a | 4421 | −50.0 | 9.9 × 10−6 | −2.26 | 5.57 × 10−2 | 0.625 | ATP-dependent RNA helicase DDX39A | XBP-1 [T00902]; E2F-1 [T01542]; f(alpha)-f(epsilon) [T00287] |
| NC_035089.1_41352276 | LOC110486510/Ddx39a | 4422 | −55.0 | 5.2 × 10−6 | −2.26 | 5.57 × 10−2 | 0.264 | ATP-dependent RNA helicase DDX39A | XBP-1 [T00902]; E2F-1 [T01542]; f(alpha)-f(epsilon) [T00287] |
| Autophagy, glycolysis, collagen metablisim, cell memberane | |||||||||
| NC_035082.1_53102690 | LOC110526208/Atg13 | −752 | −50.6 | 1.3 × 10−5 | 2.50 | 7.86 × 10−2 | −0.674 | Autophagy-related protein 13 | Pax-2.2 [T03219]; Pax-9a [T03593] Pax-9b [T03594] |
| NC_035101.1_26128247 | LOC110504991/Sesn3 | 5519 | 27.6 | 3.4 × 10−5 | 3.69 | 1.25 × 10−2 | 0.820 | Sestrin-3 | GAGA factor [T00301] |
| NC_035093.1_46301679 | LOC110494292/Pfkfb3 | 5567 | 80.3 | 6.7 × 10−16 | 4.33 | 2.86 × 10−3 | 0.725 | 6-phosphofructo-2-kinase | MCB1 [T06035]; MCB2 [T06036]; Pax-6 [T00682] |
| NC_035094.1_5494483 | LOC110495489/Col8a1 | 8739 | −55.6 | 3.5 × 10−5 | −7.93 | 1.30 × 10−4 | 0.686 | Collagen alpha-1(VIII) chain | NF-E4 [T00560]; Sp1 [T00754]; Sp1 [T00759] |
| NC_035088.1_66751403/island11919 | LOC110538176/P3h4 | 2242 | −54.8 | 7.1 × 10−7 | −3.68 | 7.35 × 10−2 | 0.283 | Prolyl 3-hydroxylase family member 4 (non-enzymatic) | USF1 [T00874]; USF1 [T00874]; c-Myc [T00140]; PHO4 [T00690]; USF-1 [T00875]; USF-1 [T00875]; HES-1 [T01649]; HES-1 [T01649] |
| NC_035082.1_40371257 | LOC110526004/Prodhb | 4547 | 46.4 | 6.4 × 10−6 | 2.74 | 1.73 × 10−2 | 0.594 | Proline dehydrogenase 1, mitochondrial | Pax-5 [T00070]; YY1 [T04970]; RXR-beta [T01349]; HES-1 [T01649]; AhR [T00018]; AhR [T01795]; JunD [T00437] |
| NC_035088.1_41298016 | LOC110537666/Gdpd5 | 6026 | −47.6 | 1.2 × 10−5 | −20.04 | 3.30 × 10−6 | 0.139 | Glycerophosphodiester phosphodiesterase domain-containing protein 5 | |
| NC_035093.1_33080772 | LOC110494010/Aqp7 | 9509 | 29.3 | 9.4 × 10−6 | 6.05 | 2.32 × 10−3 | 0.358 | Aquaporin-7 | SXR:RXR-alpha [T05670]; HES-1 [T01649] |
| NC_035105.1_28018844/island23784 | LOC110509954/Tmem151b | 5185 | 30.6 | 3.3 × 10−5 | 10.35 | 1.65 × 10−3 | 0.639 | Transmembrane protein 151B | Pax-5 [T01201]; Nkx2-1 [T00857]; GAL4 [T00302] |
| NC_035087.1_56212451 | LOC110536085/Ank1 | 5063 | −59.5 | 1.2 × 10−5 | −2.44 | 6.32 × 10−2 | 0.079 | Ankyrin-1 | NFI/CTF [T00094]; NF-AT2 [T01945]; PEA3 [T00684]; NF-AT1 [T01948]; NF-AT1 [T01944];NF-AT1 [T00550]; HNF-4alpha1 [T00372] |
| Homeobox proteins | |||||||||
| NC_035092.1_38302815 | LOC110492001/Hox-C5a | 1862 | −47.6 | 7.8 × 10−6 | −6.03 | 1.48 × 10−2 | 0.474 | Homeobox protein Hox-C5a | c-Myb [T00138] |
| NC_035092.1_38302823 | LOC110492001/Hox-C5a | 1870 | −45.7 | 2.6 × 10−5 | −6.03 | 1.48 × 10−2 | 0.382 | Homeobox protein Hox-C5a | Sp1 [T00755]; Nkx2-1 [T00857] |
| NC_035101.1_61368566 | Six4 | 2175 | −50.8 | 3.6 × 10−5 | −3.08 | 6.73 × 10−2 | 0.015 | SIX homeobox 4 | MF3 [T00507]; ENKTF-1 [T00255]; Tll [T00789]; E2F [T00221]; POU4F1(l) [T01877]; Pax-5 [T00070] |
| Other epimarkers | |||||||||
| NC_035084.1_10667325 | Hsp90ba | 6124 | 63.5 | 8.6 × 10−7 | 2.00 | 9.16 × 10−2 | 0.698 | Heat shock 90kDa protein 1 beta isoform a | TAF [T00778] |
| NC_035091.1_7350831 | LOC110489569/Ccts | 3278 | −45.2 | 3.7 × 10−6 | −2.72 | 8.21 × 10−4 | 0.297 | T-complex protein 1 subunit epsilon | Mad [T04378]; LF-A1 [T00467]; MF3 [T00507]; E2F-1 [T01543]; YY1 [T00865]; XBP-1 [T00902]; YY1 [T00915]; Sp1 [T00754]; Sp1 [T00759]; YY1 [T00278]; ENKTF-1 [T00255] |
| NC_035084.1_34778859 | LOC110529890/PGBD4 | 923 | 31.6 | 1.9 × 10−6 | −8.21 | 8.85 × 10−6 | −0.505 | PiggyBac transposable element-derived protein 4 | HNF-1B [T01950]; Crx [T03461]; POU2F1 [T00643] |
| NC_035077.1_13381573 | LOC110493215/Mast3 | 4520 | 33.3 | 1.2 × 10−5 | 2.68 | 9.8 × 10−2 | −0.119 | Microtubule-associated serine/threonine-protein kinase 3 | C/EBP [T01388]; CREMtau [T01309]; CREMtaualpha [T01602]; CREMtau1 [T02108]; CREMtau2 [T02109] |
| NC_035092.1_36914362 | LOC110491948/Cacna1sb | 2646 | −38.6 | 2.0 × 10−6 | −5.37 | 2.96 × 10−6 | 0.858 | Dihydropyridine-sensitive L-type skeletal muscle calcium channel subunit alpha-1 | MED8 [T03491]; C/EBP [T01388]; C/EBP [T01388] |
| NC_035092.1_29706361 | LOC110491830/Cytc-1 | 4423 | −43.8 | 3.3 × 10−5 | −2.80 | 1.46 × 10−4 | 0.123 | Cytochrome c1 | Mad [T04378]; GA-BF [T00297] |
| NC_035090.1_23141191 | LOC110488198/Cpsf1 | 8837 | −50.0 | 2.1 × 10−7 | −3.83 | 1.34 × 10−2 | 0.406 | Cleavage and polyadenylation specificity factor subunit 1 | Pax-5 [T00070] |
| NC_035093.1_25737498 | LOC110493184/Znfx-1 | 2285 | −29.7 | 4.8 × 10−5 | −2.61 | 7.87 × 10−2 | 0.324 | NFX1-type zinc finger-containing protein 1 | |
| NC_035097.1_35527983 | LOC110500515/Gilgyf1 | 6282 | 48.8 | 1.7 × 10−5 | 2.74 | 7.18 × 10−2 | 0.772 | GRB10-interacting GYF protein 1 | Mad [T04378]; Pax-6 [T00682] |
| NC_035095.1_15029930 | LOC110497457 | 4136 | 47.8 | 2.0 × 10−5 | 67.32 | 8.65 × 10−3 | 0.637 | Vegetative cell wall protein gp1 | |
| NC_035097.1_10910032 | LOC110501017 | 164 | 51.6 | 5.3 × 10−8 | −4.36 | 2.37 × 10−2 | −0.417 | Uncharacterized LOC110501017 | HSF1 (long) [T01042]; HSF1 (short) [T02104]; AP-4 [T00036]; myogenin [T00528]; Cutl1 [T02042]; HOXA5 [T00377]; POU1F1a [T00691];PF1 [T04784] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, M.; Al-Tobasei, R.; Ali, A.; Kenney, B. Integrated Analyses of DNA Methylation and Gene Expression of Rainbow Trout Muscle under Variable Ploidy and Muscle Atrophy Conditions. Genes 2022, 13, 1151. https://doi.org/10.3390/genes13071151
Salem M, Al-Tobasei R, Ali A, Kenney B. Integrated Analyses of DNA Methylation and Gene Expression of Rainbow Trout Muscle under Variable Ploidy and Muscle Atrophy Conditions. Genes. 2022; 13(7):1151. https://doi.org/10.3390/genes13071151
Chicago/Turabian StyleSalem, Mohamed, Rafet Al-Tobasei, Ali Ali, and Brett Kenney. 2022. "Integrated Analyses of DNA Methylation and Gene Expression of Rainbow Trout Muscle under Variable Ploidy and Muscle Atrophy Conditions" Genes 13, no. 7: 1151. https://doi.org/10.3390/genes13071151
APA StyleSalem, M., Al-Tobasei, R., Ali, A., & Kenney, B. (2022). Integrated Analyses of DNA Methylation and Gene Expression of Rainbow Trout Muscle under Variable Ploidy and Muscle Atrophy Conditions. Genes, 13(7), 1151. https://doi.org/10.3390/genes13071151