
The First Complete Mitochondrial Genome of Eucrate crenata (Decapoda: Brachyura: Goneplacidae) and Phylogenetic Relationships within Infraorder Brachyura




Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Identification and DNA Extraction
2.2. Illumina Sequencing, Mitogenome Assembly and Annotation
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
2.5. Positive Selection Analysis
3. Results and Discussion
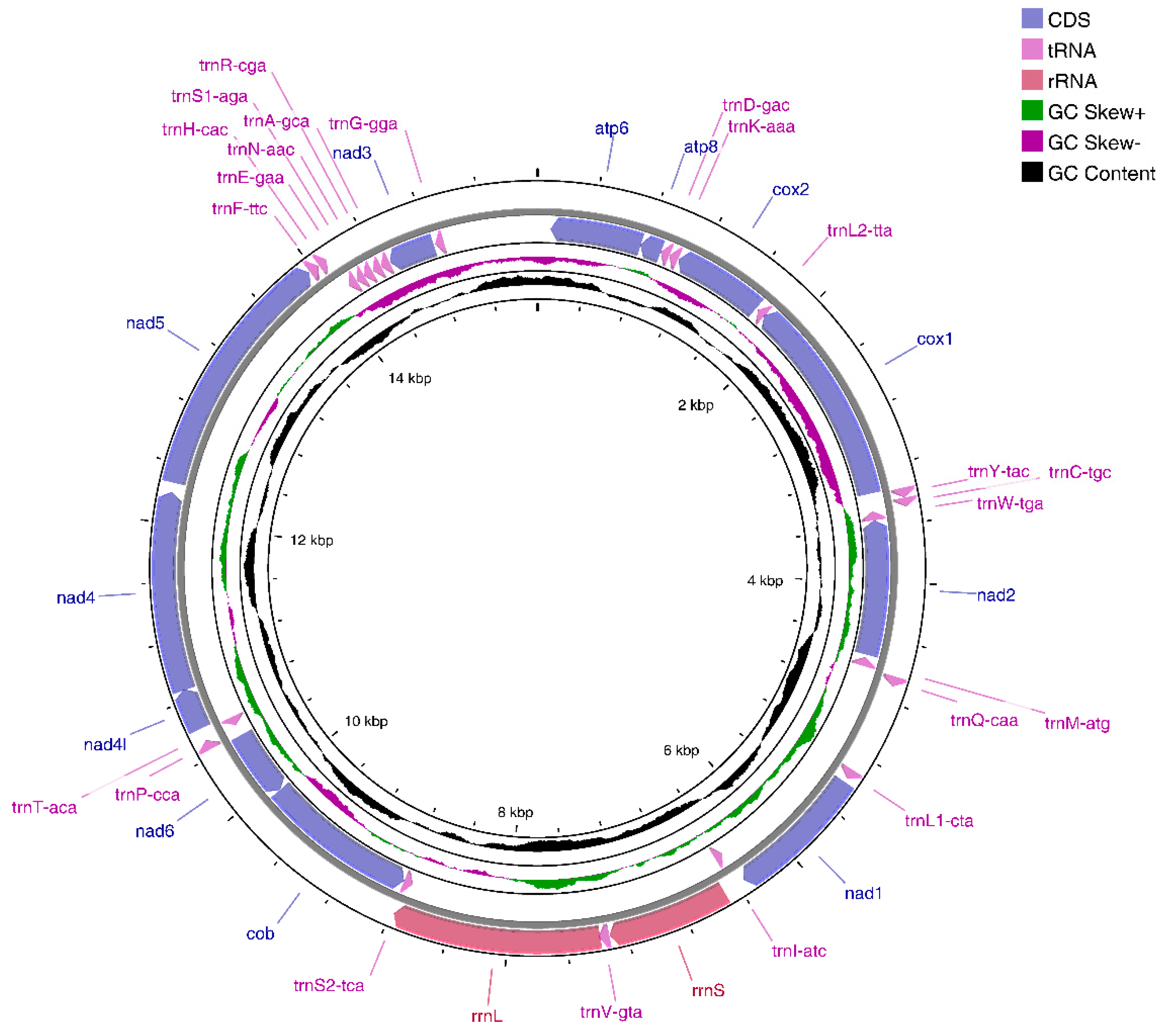
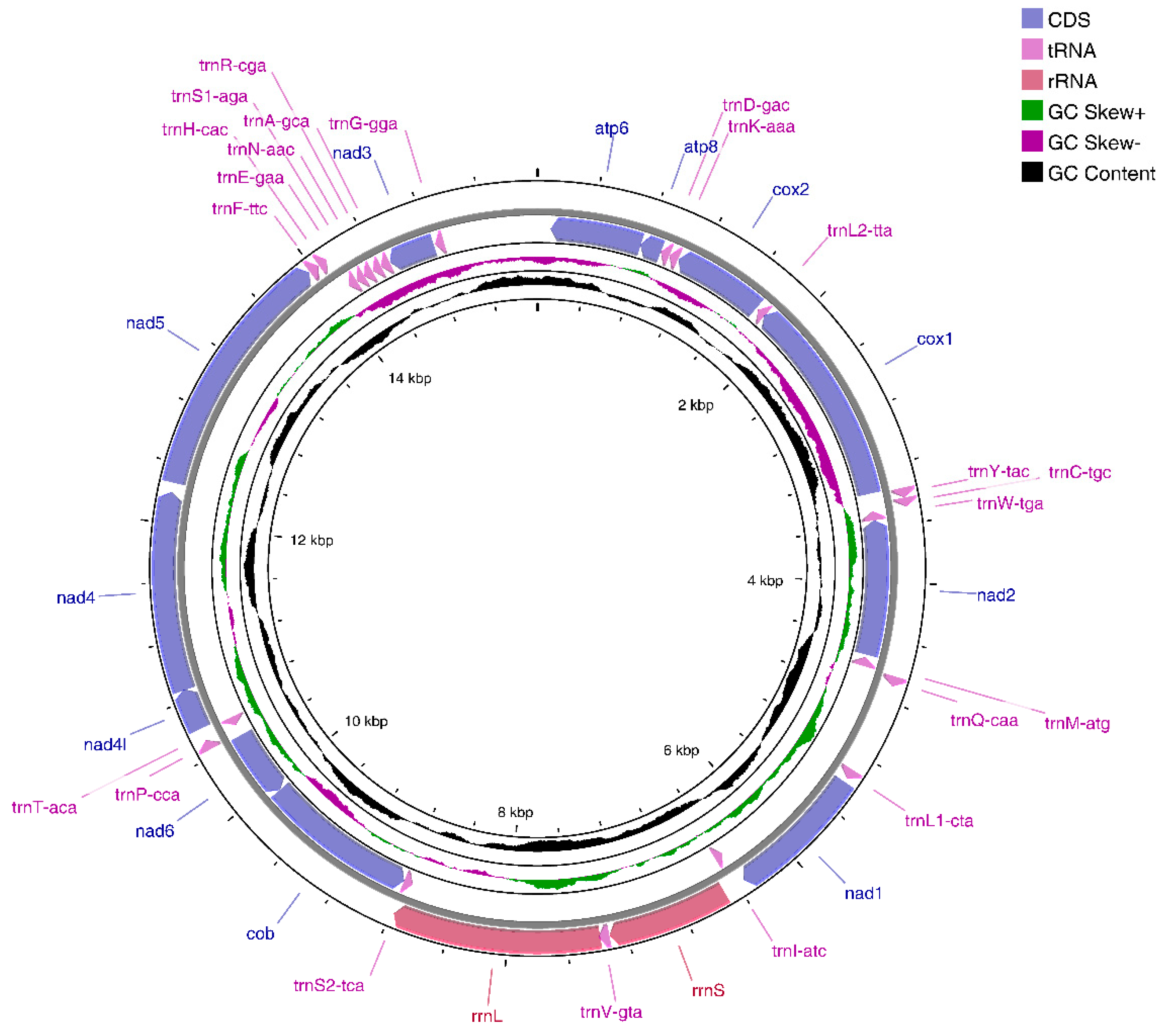
3.1. E. crenata Mitogenome Organization
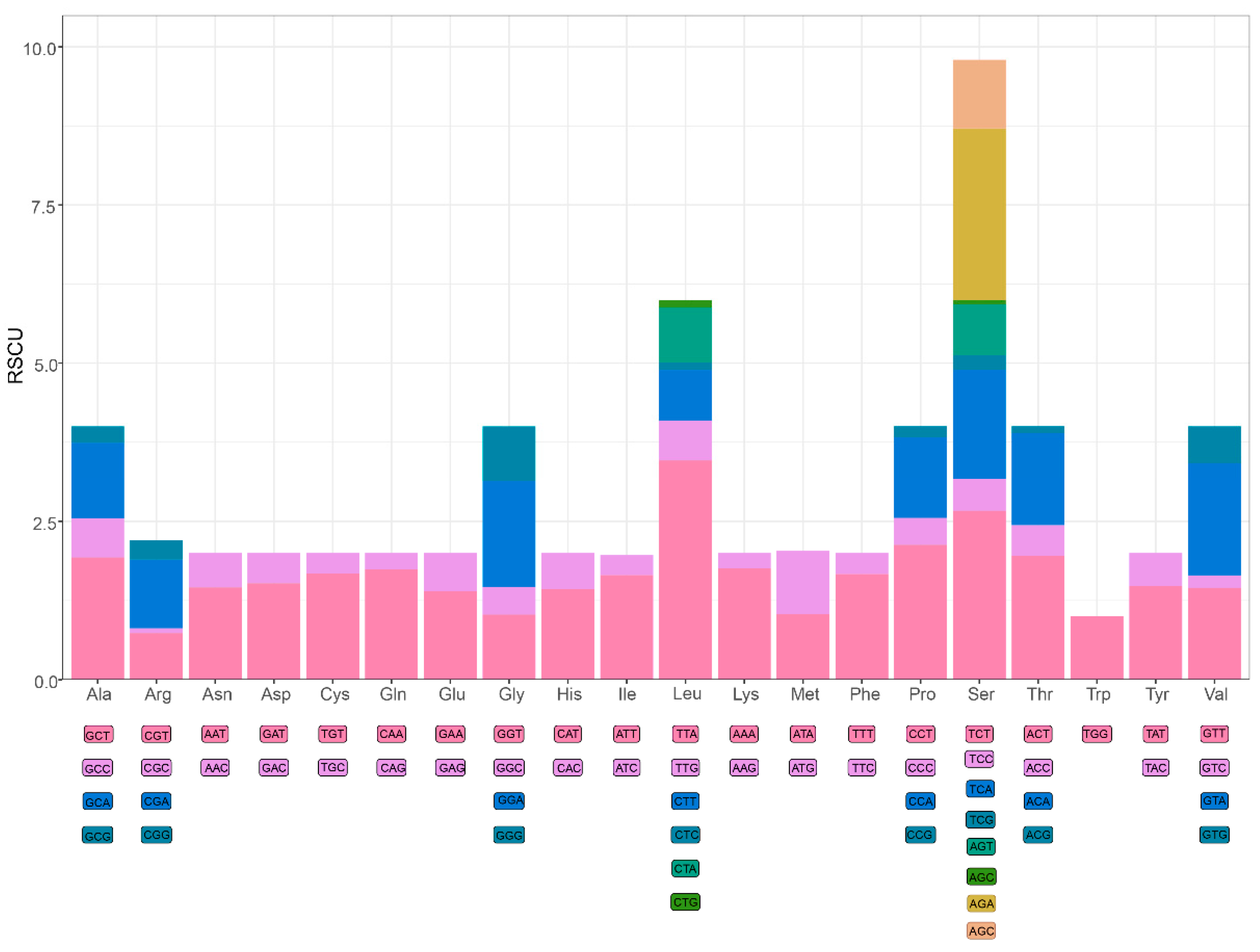
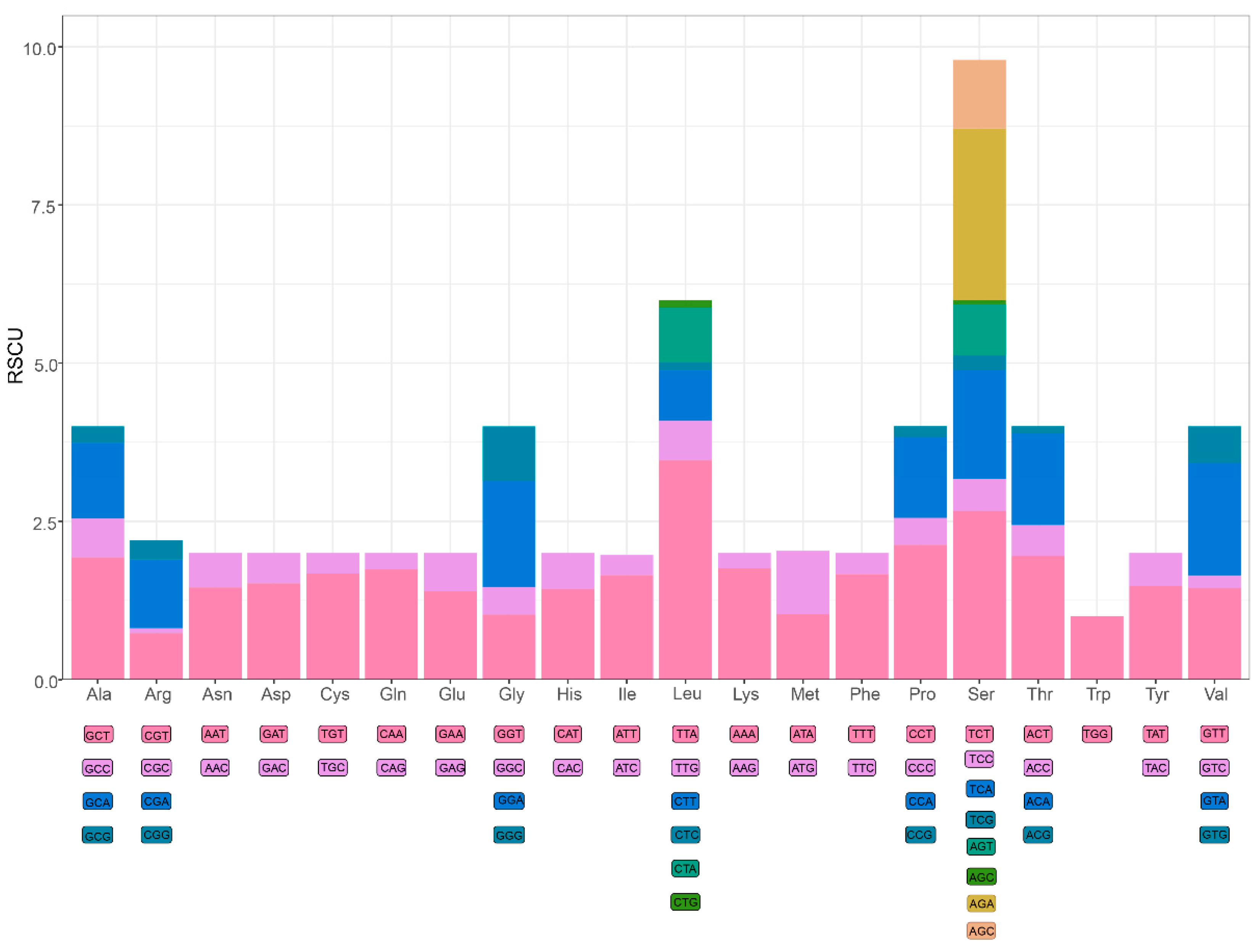
3.2. Protein-Coding Genes
3.3. Ribosomal RNA and Transfer RNA Genes
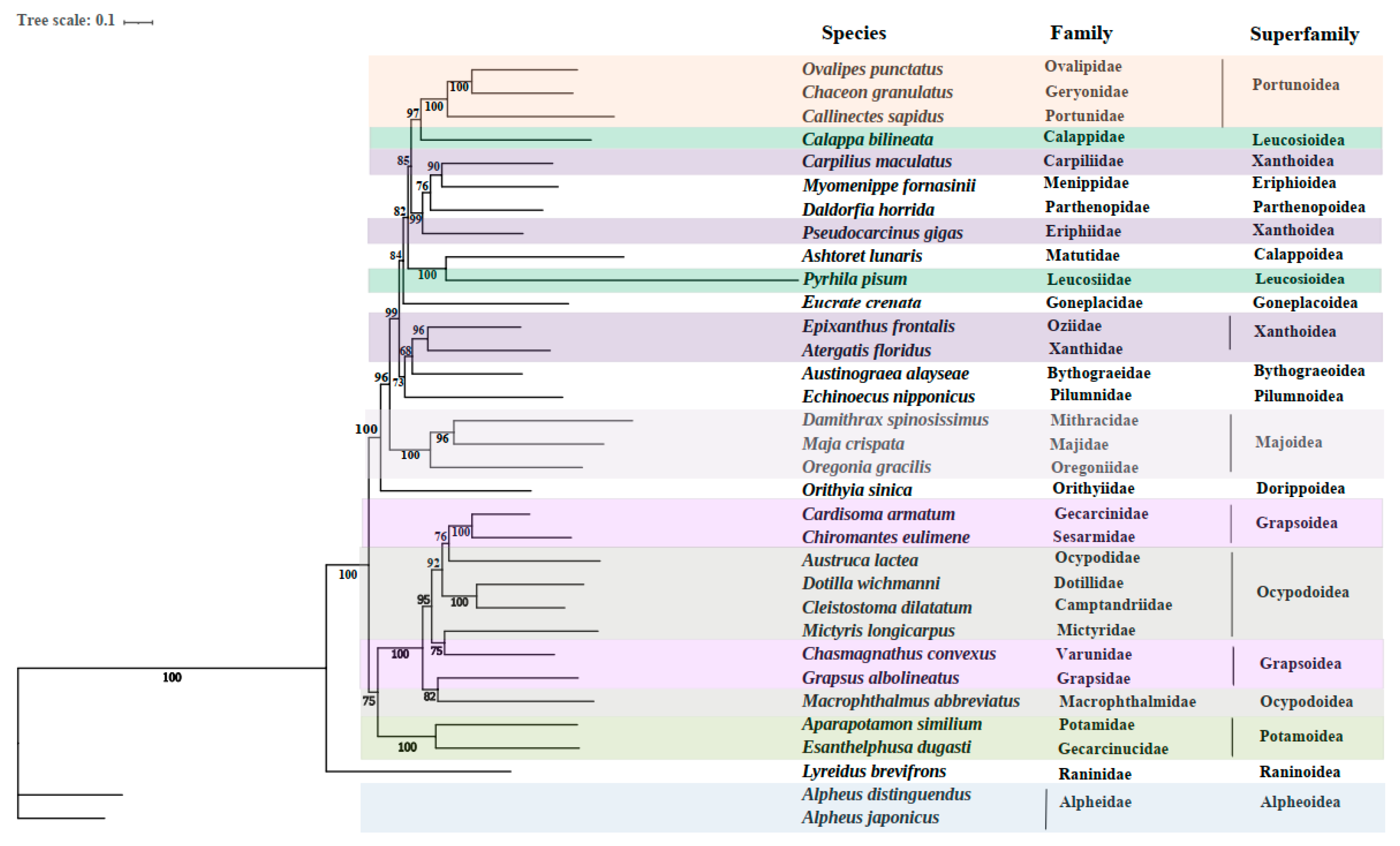
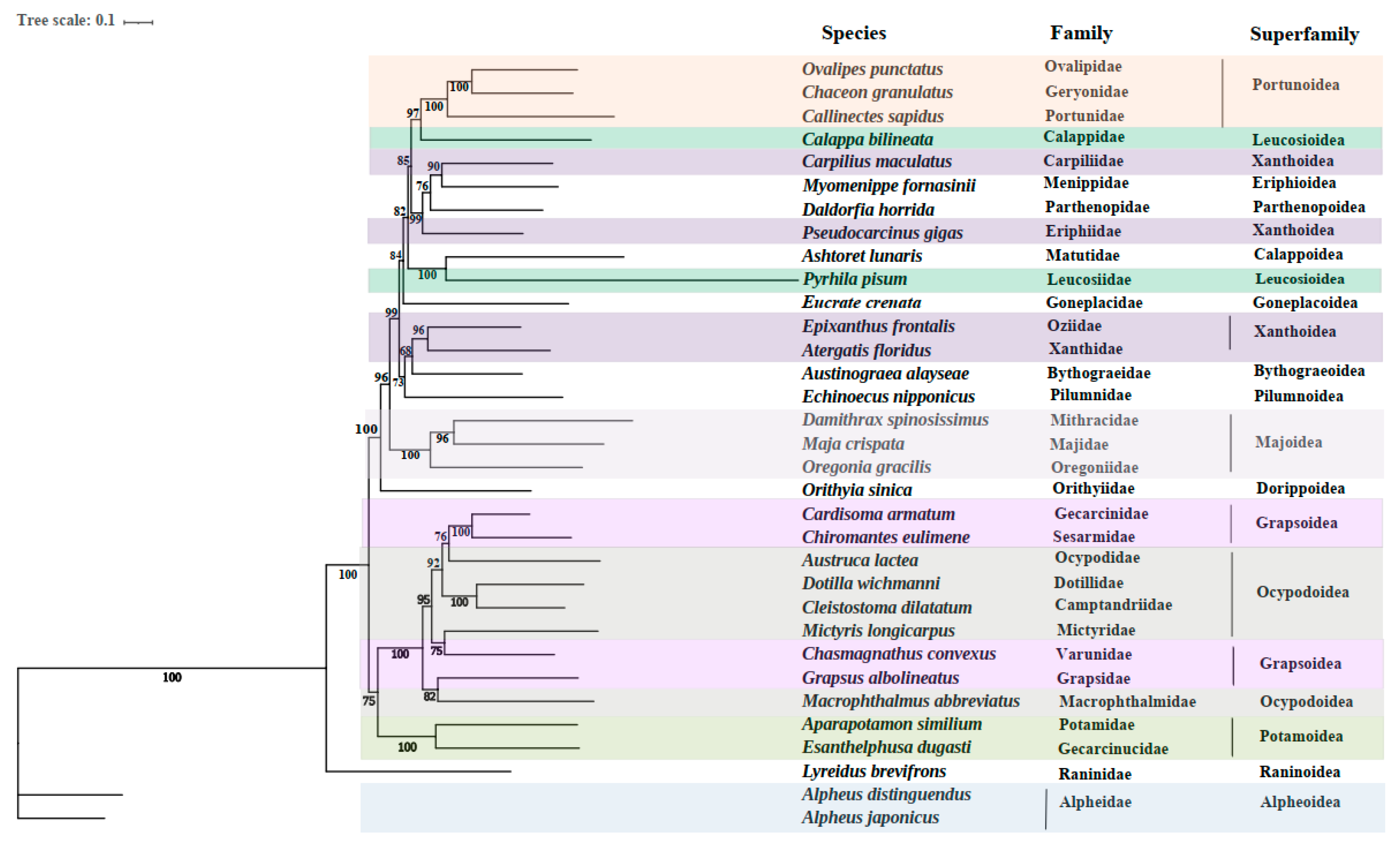
3.4. Phylogenetic Relationships
3.5. Positive Selection Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Boore, J.L.; Macey, J.R.; Medina, M. Sequencing and comparing whole mitochondrial genomes of animals. Method Enzymol. 2005, 395, 311–348. [Google Scholar]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, S.; Kumar, V.; Tyagi, K.; Chakraborty, R.; Chandra, K. The first complete mitochondrial genome of the Indian tent turtle, Pangshura tentoria (Testudines: Geoemydidae): Characterization and comparative analysis. Ecol. Evol. 2019, 9, 10854–10868. [Google Scholar] [CrossRef] [Green Version]
- Dhar, D.; Dey, D.; Basu, S.; Fortunato, H. Understanding the adaptive evolution of mitochondrial genomes in intertidal chitons. BioRxiv 2020, 3, 980664. [Google Scholar]
- Tan, M.H.; Gan, H.M.; Lee, Y.P.; Linton, S.; Grandjean, F.; Bartholomei-Santos, M.L.; Miller, A.D.; Austin, C.M. ORDER within the chaos: Insights into phylogenetic relationships within the Anomura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Mol. Phylogenet. Evol. 2018, 127, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.; Vargas, S.; Knapp, I.S.; Pomponi, S.A.; Toonen, R.J.; Erpenbeck, D.; Worheide, G. Divergence times in demosponges (Porifera): First insights from new mitogenomes and the inclusion of fossils in a birthdeath clock model. BMC Evol. Biol. 2018, 18, 114. [Google Scholar] [CrossRef]
- Basso, A.; Babbucci, M.; Pauletto, M.; Riginella, E.; Patarnello, T.; Negrisolo, E. The highly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Sci. Rep. 2017, 7, 4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.K.; Wang, A.; Lu, X.L.; Song, D.H.; Jin, Y.H.; Lu, J.J.; Sun, H.Y. Mitochondrial genomes of two Brachyuran crabs (Crustacea: Decapoda) and phylogenetic analysis. J. Crustacean Biol. 2014, 34, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Castro, P. A reappraisal of the family Goneplacidae MacLeay, 1838 (Crustacea, Decapoda, Brachyura) and revision of the subfamily Goneplacinae, with the description of 10 new genera and 18 new species. Zoosystema 2007, 29, 609–774. [Google Scholar]
- Karasawa, H.; Kato, H. The systematic status of the genus Miosesarma Karasawa, 1989 with a phylogenetic analysis within the family Grapsidae and a review of fossil records (Crustacea: Decapoda: Brachyura). Paleontol. Res. 2001, 5, 259–275. [Google Scholar]
- Ng, P.K.L.; Castro, P. A revision of Carcinoplax abyssicola (Miers, 1885) and seven related species of Carcinoplax, H. Milne Edwards, 1852, with the description of two new species and an updated key to the genus (Crustacea, Decapoda, Brachyura, Goneplacidae). Zoosystema 2020, 42, 239–284. [Google Scholar] [CrossRef]
- Lu, J.Y.; Xia, L.P.; Liu, X.J.; Ma, Y.W.; Li, J.J.; Ye, Y.Y.; Guo, B.Y. The mitochondrial genome of Grapsus albolineatus (Decapoda: Brachyura: Grapsidae) and phylogenetic associations in Brachyura. Sci. Rep. 2022, 12, 2104. [Google Scholar] [CrossRef]
- Rodriguez-Pilco, M.A.; Lesny, P.; Podsiadlowski, L.; Schubart, C.D.; Baeza, J.A. Characterization of the Complete Mitochondrial Genome of the Bromeliad Crab Metopaulias depressus (Rathbun, 1896) (Crustacea: Decapoda: Brachyura: Sesarmidae). Genes 2022, 13, 299. [Google Scholar] [CrossRef]
- Jennings, L.A.; Blakeslee, A.M.H.; McCoy, K.A.; Behringer, D.C.; Bojko, J. Systematic assessment of the Panopeidae and broader Eubrachyura (Decapoda: Brachyura) using mitochondrial genomics. Arthropod Syst. Phylo. 2021, 79, 569–585. [Google Scholar] [CrossRef]
- Nayak, A.; Roy, M.K.D.; Mohanty, B.; Rout, S.S.; Dash, B.; Raman, A.V.; Patnaik, L.; Raut, D. On the occurrence of an Euryplacid crab Eucrate crenata (De Haan, 1835), a first record from Odisha, India. Indian J. Geo-Mar. Sci. 2020, 49, 1105–1107. [Google Scholar]
- Lim, H.J.; Lee, E.H.; Yoon, Y.; Chua, B.; Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. S. 1992, 23, 263–286. [Google Scholar] [CrossRef]
- Yang, Z.H. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4, a program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Nielsen, R.; Goldman, N.; Pedersen, A.M.K. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Nielsen, R.; Yang, Z.H. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef]
- Yang, Z.H.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.D.; Wu, X.Y.; Yu, Z.N. The mitogenome of Paphia euglypta (Bivalvia: Veneridae) and comparative mitogenomic analyses of three venerids. Genome 2010, 53, 1041–1052. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, K.Y.; Lee, S.H.; Hong, S.S.; Cho, I.Y.; Yi, C.H.; Kim, I.H.; Yoon, M.; Kim, M.S. The complete mitochondrial genome of Pseudohelice subquadrata (Dana, 1851) (Crustacea: Decapoda: Varunidae). Mitochondrial DNA B 2019, 4, 103–104. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tang, D.; Guo, H.Y.; Wang, J.; Xu, X.Y.; Wang, Z.F. Comparative mitochondrial genomic analysis of Macrophthalmus pacificus and insights into the phylogeny of the Ocypodoidea & Grapsoidea. Genomics 2020, 112, 82–91. [Google Scholar]
- Karagozlu, M.Z.; Kim, J.I.; Choi, T.J.; Dinh, T.D.; Kim, C.B. The complete mitochondrial genome of Hemigrapsus penicillatus (De Haan, 1835) (Decapoda, Varunidae). Mitochondrial DNA B 2018, 3, 261–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.M.; Lee, J.; Kim, S.K.; Karagozlu, M.Z.; Kim, C.B. The complete mitochondrial genome of Grapsus tenuicrustatus (Herbst, 1783) (Decapoda, Grapsidae). Mitochondrial DNA B 2016, 1, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Wang, Z.Q.; Shi, X.J.; Wu, Q.; Tao, Y.T.; Guo, H.Y.; Ji, C.Y.; Bai, Y.Z. Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int. J. Biol. Macromol. 2018, 118, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gong, L.; Lu, X.T.; Jiang, L.H.; Liu, B.J.; Liu, L.Q.; Lu, Z.M.; Li, P.F.; Zhang, X. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 2020, 162, 704–714. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, K.H.; Liu, Y.F.; Zhang, H.; Gong, L.; Jiang, L.H.; Liu, L.Q.; Lu, Z.M.; Liu, B.J. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 2021, 11, 2411. [Google Scholar] [CrossRef]
- Gong, L.; Lu, X.T.; Wang, Z.F.; Zhu, K.H.; Liu, L.Q.; Jiang, L.H.; Lu, Z.M.; Liu, B.J. Novel gene rearrangement in the mitochondrial genome of Coenobita brevimanus (Anomura: Coenobitidae) and phylogenetic implications for Anomura. Genomics 2020, 112, 1804–1812. [Google Scholar] [CrossRef]
- Park, Y.J.; Park, C.E.; Lee, S.H.; Ko, H.S.; Ullah, I.; Hwang, U.W.; Shin, J.H. The complete mitochondrial genome sequence of the intertidal crab Parasesarma tripectinis (Arthropoda, Decapoda, Sesarmidae). Mitochondrial DNA B 2018, 3, 193–194. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.T.; Xin, Z.Z.; Tang, Y.Y.; Yang, T.T.; Tang, B.P.; Sun, Y.; Zhang, D.Z.; Zhou, C.L.; Liu, Q.N.; Yu, X.M. Comparative mitochondrial genome analyses of sesarmid and other brachyuran crabs reveal gene rearrangements and phylogeny. Front. Genet. 2020, 11, 536640. [Google Scholar] [CrossRef]
- Kennish, R.; Williams, G.A. Feeding preferences of the herbivorous crab Grapsus albolineatus: The differential influence of algal nutrient content and morphology. Mar. Ecol. Prog. Ser. 1997, 147, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.Y.; Liu, X.M.; Lin, F.; Xie, Z.F.; Fazhan, H.; Ikhwanuddin, M.; Tan, H.Q.; Ma, H.Y. The whole mitochondrial genome of the mangrove crab, Metopograpsus frontalis (Miers, 1880) (Decapoda, Grapsidae) and its phylogenetic relationship. Mitochondrial DNA B 2018, 3, 368–369. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Shi, X.J.; Guo, H.Y.; Tang, D.; Bai, Y.Z.; Wang, Z.F. Characterization of the complete mitochondrial genome of Uca lacteus and comparison with other Brachyuran crabs. Genomics 2020, 112, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.Y.; Gan, H.M.; Tan, M.H.; Lee, Y.P.; Austin, C.M. The complete mitogenome of the hermit crab Clibanarius infraspinatus (Hilgendorf, 1869) (Crustacea: Decapoda: Diogenidae)—A new gene order for the Decapoda. Mitochondrial DNA A 2016, 27, 4099–4100. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.P.; Xin, Z.Z.; Liu, Y.; Zhang, D.Z.; Wang, Z.F.; Zhang, H.B.; Chai, X.Y.; Zhou, C.L.; Liu, Q.N. The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS ONE 2017, 12, e0179800. [Google Scholar]
- Yang, J.S.; Nagasawa, H.; Fujiwara, Y.; Tsuchida, S.; Yang, W.J. The complete mitogenome of the hydrothermal vent crab Gandalfus yunohana (Crustacea: Decapoda: Brachyura): A link between the Bythograeoidea and Xanthoidea. Zool. Scr. 2010, 39, 621–630. [Google Scholar] [CrossRef]
- Xing, Y.H.; Ma, X.P.; Wei, Y.Q.; Pan, D.; Liu, W.L.; Sun, H.Y. The complete mitochondrial genome of the semiterrestrial crab, Chiromantes neglectum (Eubrachyura: Grapsoidea: Sesarmidae). Mitochondrial DNA B 2016, 1, 461–463. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.F.; Shi, X.J.; Tao, Y.T.; Wu, Q.; Bai, Y.Z.; Guo, H.Y.; Tang, D. The complete mitochondrial genome of Parasesarma pictum (Brachyura: Grapsoidea: Sesarmidae) and comparison with other Brachyuran crabs. Genomics 2019, 111, 799–807. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Gong, L.; Lu, X.T.; Jiang, L.H.; Liu, B.J.; Liu, L.Q.; Lu, Z.M.; Li, P.F. Mitochondrial Genome of Episesarma lafondii (Brachyura: Sesarmidae) and Comparison with Other Sesarmid Crabs. J. Ocean. U. China 2021, 20, 1545–1556. [Google Scholar] [CrossRef]
- Baeza, J.A. The complete mitochondrial genome of the Caribbean spiny lobster Panulirus argus. Sci. Rep. 2018, 8, 17690. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. TRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Dreyer, H.; Steiner, G. The complete sequences and gene organisation of the mitochondrial genome of the heterodont bivalves Acanthocardia tuberculata and Hiatella arctica–and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front. Zool. 2006, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genom. 2008, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Li, Q. Complete mitochondrial DNA sequence of Crassostrea nippona: Comparative and phylogenomic studies on seven commercial Crassostrea species. Mol. Biol. Rep. 2012, 39, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Li, C.; Fang, W.Y.; Yu, X.P. The complete mitochondrial genome of two Tetragnatha Spiders (Araneae: Tetragnathidae): Severe truncation of tRNAs and novel gene rearrangements in araneae. Int. J. Biol. Sci. 2016, 12, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.E.; Sha, Z.L.; Wang, Y.R. Complete mitochondrial genome of the first deep-sea spongicolid shrimp Spongiocaris panglao (Decapoda: Stenopodidea): Novel gene arrangement and the phylogenetic position and origin of Stenopodidea. Gene 2018, 676, 123–138. [Google Scholar] [CrossRef]
- Chai, H.N.; Du, Y.Z.; Zhai, B.P. Characterization of the complete mitochondrial genome of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci. 2012, 8, 561–579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, Y.H.; Wang, X.; Zhang, H.X.; Lin, Q. The mitochondrial genome of a sea anemone Bolocera sp. exhibits novel genetic structures potentially involved in adaptation to the deep-sea environment. Ecol. Evol. 2017, 7, 4951–4962. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.E.; Hui, M.; Wang, M.X.; Sha, Z.L. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Phys. D 2018, 25, 42–52. [Google Scholar] [CrossRef]
- Tan, M.H.; Gan, H.M.; Lee, Y.P.; Austin, C.M. The complete mitogenome of the moon crab Ashtoret lunaris (Forskal, 1775), (Crustacea; Decapoda; Matutidae). Mitochondrial DNA Part A 2016, 27, 1313–1314. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Liang, L.; Zhu, Z.H.; Zhou, W.P.; Irwin, D.M.; Zhang, Y.P. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [Green Version]
- Tomasco, I.H.; Lessa, E.P. The evolution of mitochondrial genomes in subterranean caviomorph rodents: Adaptation against a background of purifying selection. Mol. Phylogenet. Evol. 2011, 61, 64–70. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.X.; Li, X.Z. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Fonseca, R.R.; Johnson, W.E.; O’Brien, S.J.; Ramos, M.J.; Antunes, A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genom. 2008, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.L.; Dong, X.Q.; Shi, L.; Shi, L.; Lin, K.Q.; Huang, X.Q.; Chu, J.Y. Differences in mtDNA whole sequence between Tibetan and Han populations suggesting adaptive selection to high altitude. Gene 2012, 496, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Xu, S.X.; Xu, J.X.; Guo, Y.; Yang, G. Adaptive evolution of mitochondrial energy metabolism genes associated with increased energy demand in flying insects. PLoS ONE 2014, 9, e99120. [Google Scholar] [CrossRef] [Green Version]
- Plazzi, F.; Puccio, G.; Passamonti, M. Burrowers from the past: Mitochondrial signatures of Ordovician bivalve infaunalization. Genome Biol. Evol. 2017, 9, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Nielsen, R.; Yang, Z.H. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.J.; Gao, W.X.; Gao, Y.Q.; Tang, S.; Huang, Q.Y.; Tan, X.L.; Chen, J.; Huang, T.S. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion 2008, 8, 352–357. [Google Scholar] [CrossRef]
- Mahalingam, S.; McClelland, G.B.; Scott, G.R. Evolved changes in the intracellular distribution and physiology of muscle mitochondria in high-altitude native deer mice. J. Physiol. 2017, 595, 4785–4801. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Dirmeier, R.; O’Brien, K.M.; Engle, M.; Dodd, A.; Spears, E.; Poyton, R.O. Exposure of yeast cells to anoxia induces transient oxidative stress-Implications for the induction of hypoxic genes. J. Biol. Chem. 2002, 277, 34773–34784. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.D.; Liu, J.; Zhang, H.B. Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS ONE 2018, 13, e0208051. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.D.; Liu, J.; Zhang, H.B. The first complete mitochondrial genome of the Mariana Trench Freyastera benthophila (Asteroidea: Brisingida: Brisingidae) allows insights into the deep-sea adaptive evolution of Brisingida. Ecol. Evol. 2018, 8, 10673–10686. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Species | Size (bp) | Accession. no |
|---|---|---|---|---|
| Bythograeoidea | Bythograeidae | Austinograea alayseae | 15,620 | NC_020314 |
| Calappoidea | Matutidae | Ashtoret lunaris | 15,807 | NC_024435 |
| Dorippoidea | Orithyiidae | Orithyia sinica | 15,568 | NC_039639 |
| Eriphioidea | Menippidae | Myomenippe fornasinii | 15,658 | NC_024437 |
| Goneplacoidea | Goneplacidae | Eucrate crenata | 15,597 | ON150678 |
| Leucosioidea | Calappidae | Calappa bilineata | 15,606 | NC_047195 |
| Leucosiidae | Pyrhila pisum | 15,516 | NC_030047 | |
| Majoidea | Majidae | Maja crispate | 16,592 | NC_035424 |
| Mithracidae | Damithrax spinosissimus | 15,817 | NC_025518 | |
| Oregoniidae | Oregonia gracilis | 15,737 | NC_057204 | |
| Parthenopoidea | Parthenopidae | Daldorfia horrida | 15,737 | NC_049029 |
| Pilumnoidea | Pilumnidae | Echinoecus nipponicus | 16,173 | NC_039618 |
| Portunoidea | Geryonidae | Chaceon granulatus | 16,135 | NC_023476 |
| Ovalipidae | Ovalipes punctatus | 16,084 | NC_042695 | |
| Portunidae | Callinectes sapidus | 16,263 | NC_006281 | |
| Potamoidea | Gecarcinucidae | Esanthelphusa dugasti | 19,437 | NC_060554 |
| Potamidae | Aparapotamon similium | 19,236 | NC_053821 | |
| Xanthoidea | Carpiliidae | Carpilius maculatus | 15,761 | NC_049030 |
| Eriphiidae | Pseudocarcinus gigas | 15,515 | NC_006891 | |
| Oziidae | Epixanthus frontalis | 15,993 | NC_039110 | |
| Xanthidae | Atergatis floridus | 16,180 | NC_037201 | |
| Raninoidea | Raninidae | Lyreidus brevifrons | 16,112 | NC_026721 |
| Grapsoidea | Gecarcinidae | Cardisoma armatum | 15,586 | NC_057477 |
| Grapsidae | Grapsus albolineatus | 15,578 | NC_057301 | |
| Sesarmidae | Chiromantes eulimene | 15,894 | NC_047209 | |
| Varunidae | Chasmagnathus convexus | 15,107 | NC_052834 | |
| Ocypodoidea | Camptandriidae | Cleistostoma dilatatum | 15,444 | NC_060620 |
| Dotillidae | Dotilla wichmanni | 15,600 | NC_038180 | |
| Macrophthalmidae | Macrophthalmus abbreviatus | 16,322 | NC_057472 | |
| Mictyridae | Mictyris longicarpus | 15,548 | NC_025325 | |
| Ocypodidae | Austruca lactea | 15,659 | NC_042401 | |
| Outgroup | Alpheidae | Alpheus distinguendus | 15,700 | NC_014883 |
| Alpheus japonicus | 16,619 | NC_038116 |
| Gene | Strand | Position | Length (bp) | Intergenic_spacer | Start_coden | Stop_coden | Anticodon |
|---|---|---|---|---|---|---|---|
| cox3 | R | 1–96 | 96 | - | - | - | |
| atp6 | R | 96–767 | 672 | −1 | ATA | TAA | |
| atp8 | R | 764–922 | 159 | −4 | ATG | TAG | |
| trnD-gac | R | 923–990 | 68 | 0 | - | - | GAC |
| trnK-aaa | R | 991–1059 | 69 | 0 | - | - | AAA |
| cox2 | R | 1064–1735 | 672 | 4 | ATA | TAA | |
| trnL2-tta | R | 1754–1818 | 65 | 18 | - | - | UUA |
| cox1 | R | 1814–3352 | 1539 | −5 | ATG | TAA | |
| trnY-tac | F | 3362–3427 | 66 | 9 | - | - | UAC |
| trnC-tgc | F | 3428–3489 | 62 | 0 | - | - | UGC |
| trnW-tga | R | 3482–3549 | 68 | −8 | - | - | UGA |
| nad2 | R | 3548–4537 | 990 | −2 | ATA | TAG | |
| trnM-atg | R | 4556–4623 | 68 | 18 | - | - | AUG |
| trnQ-caa | F | 4630–4698 | 69 | 6 | - | - | CAA |
| trnL1-cta | F | 5313–5380 | 68 | 614 | - | - | CUA |
| nad1 | F | 5409–6347 | 939 | 28 | ATA | TAA | |
| trnI-atc | R | 6371–6436 | 66 | 23 | - | - | AUC |
| rrnS | F | 6500–7318 | 819 | 63 | - | - | |
| trnV-gta | F | 7320–7392 | 73 | 1 | - | - | GUA |
| rrnL | F | 7381–8778 | 1398 | −12 | - | - | |
| trnS2-tca | R | 8733–8800 | 68 | −46 | - | - | UCA |
| cob | R | >8801–9935 | 1135 | 0 | ATG | T | |
| nad6 | R | 9935–10,426 | 492 | −1 | ATT | TAA | |
| trnP-cca | F | 10,444–10,510 | 67 | 17 | - | - | CCA |
| trnT-aca | R | 10,511–10,574 | 64 | 0 | - | - | ACA |
| nad4l | F | 10,595–10,879 | 285 | 20 | ATG | TAA | |
| nad4 | F | 10,873–12,207 | 1335 | −7 | ATG | TAA | |
| nad5 | F | 12,271–13,965 | 1695 | 63 | ATA | TAA | |
| trnF-ttc | F | 13,972–14,039 | 68 | 6 | - | - | UUC |
| trnH-cac | F | 14,046–14,109 | 64 | 6 | - | - | CAC |
| trnE-gaa | R | 14,136–14,203 | 68 | 26 | - | - | GAA |
| trnS1-aga | R | 14,204–14,269 | 66 | 0 | - | - | AGA |
| trnN-aac | R | 14,270–14,337 | 68 | 0 | - | - | AAC |
| trnR-cga | R | 14,344–14,409 | 66 | 6 | - | - | CGA |
| trnA-gca | R | 14,411–14,478 | 68 | 1 | - | - | GCA |
| nad3 | R | 14,482–14,826 | 345 | 3 | ATT | TAA | |
| trnG-gga | R | 14,836–14,903 | 68 | 9 | - | - | GGA |
| Trees | Models | lnL | Parameter Estimates | Model Compared | 2ΔL | LRT p-Value |
|---|---|---|---|---|---|---|
| Branch models | ||||||
| ML tree | M0 | −211,767.521149 | ω = 0.04109 | |||
| Two-ratio | −211,744.724139 | ω0 = 0.04084 ω1 = 999.00000 | Two-ratio vs. M0 | 45.5940200000186 | 0 | |
| Free-tatio | −210,762.85387 | Free-ratio vs. M0 | 2009.33455800003 | 0 | ||
| Branch-sits models | ||||||
| ML tree | Null model | −207,044.061032 | p0 = 0.76754; p1 = 0.09376; p2a = 0.12361; p2b = 0.01510 | |||
| ω0 = 0.03453; ω1 = 1.00000; ω2a = 1.00000; ω2b = 1.00000 | ||||||
| Model A | −207,042.434633 | p0 = 0.87845; p1 = 0.10727; p2a = 0.01272; p2b = 0.00155 | Model A vs. null model | 3.25279800000134 | 0.071301646 | |
| ω0 = 0.03452; ω1 = 1.00000; ω2a = 20.67481; ω2b = 20.67481 |
| ML Tree | |||
|---|---|---|---|
| Gene | Codon | Amino Acid | BEB Values |
| atp6 | 607 | L | 0.884 |
| cox1 | 1389 | V | 0.984 * |
| cox1 | 1459 | S | 0.888 |
| cox1 | 1469 | T | 0.887 |
| cox1 | 1828 | V | 0.876 |
| cox1 | 1961 | L | 0.829 |
| cox1 | 2015 | A | 0.876 |
| cox1 | 2021 | T | 0.512 |
| cox1 | 2033 | S | 0.826 |
| cox2 | 2344 | M | 0.86 |
| cox2 | 2411 | S | 0.769 |
| cox2 | 2611 | A | 0.863 |
| cox2 | 2955 | S | 0.908 |
| cox3 | 3274 | L | 0.632 |
| cox3 | 3308 | F | 0.886 |
| cox3 | 3469 | V | 0.569 |
| cox3 | 3475 | L | 0.535 |
| cox3 | 3511 | L | 0.534 |
| cox3 | 3515 | S | 0.841 |
| cox3 | 3605 | S | 0.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.; Han, C.; Guo, B.; Liu, K.; Lin, X.; Lu, X. The First Complete Mitochondrial Genome of Eucrate crenata (Decapoda: Brachyura: Goneplacidae) and Phylogenetic Relationships within Infraorder Brachyura. Genes 2022, 13, 1127. https://doi.org/10.3390/genes13071127
Pang X, Han C, Guo B, Liu K, Lin X, Lu X. The First Complete Mitochondrial Genome of Eucrate crenata (Decapoda: Brachyura: Goneplacidae) and Phylogenetic Relationships within Infraorder Brachyura. Genes. 2022; 13(7):1127. https://doi.org/10.3390/genes13071127
Chicago/Turabian StylePang, Xiaoke, Chenglong Han, Biao Guo, Kefeng Liu, Xiaolong Lin, and Xueqiang Lu. 2022. "The First Complete Mitochondrial Genome of Eucrate crenata (Decapoda: Brachyura: Goneplacidae) and Phylogenetic Relationships within Infraorder Brachyura" Genes 13, no. 7: 1127. https://doi.org/10.3390/genes13071127
APA StylePang, X., Han, C., Guo, B., Liu, K., Lin, X., & Lu, X. (2022). The First Complete Mitochondrial Genome of Eucrate crenata (Decapoda: Brachyura: Goneplacidae) and Phylogenetic Relationships within Infraorder Brachyura. Genes, 13(7), 1127. https://doi.org/10.3390/genes13071127