1. Introduction
Halting cancer cells’ proliferation has always been a tough challenge, because its main difficulties are related to the high molecular heterogeneity of tumors and their altered genetic and epigenetic background. In particular, cancer cells frequently show an impairment of the pathways that normally arrest cell proliferation in the presence of DNA damage, genetic alterations, and genomic instability [
1]; thus, forcing the reactivation of these pathways, such as those leading to cellular senescence, could be a valid strategy to counteract cancer cell proliferation.
Cellular senescence is defined as the irreversible arrest of the cell cycle [
2]. It was originally described by Leonard Hayflick, who noticed that cultured normal cells reach a limit on cell divisions. This limited capacity to divide depends on telomere length, which shortens with every cell division. Telomere shortening, in turn, activates a persistent DNA damage response, which leads to permanent cell-cycle arrest, known as cell senescence [
3,
4]. The activation of such a response avoids the propagation of DNA/chromosome damage and preserves the genetic information. In addition, other stress signals (ROS, radiations, other DNA damages, and oncogenes activation), both endogenous and exogenous, can trigger cellular senescence; this, in turn, acts as a protective mechanism to avoid the spread of altered cells [
5].
A senescent cell does not divide, but it is still metabolically active. It synthesizes and produces mediators that make it highly recognizable and that serve to maintain and propagate the senescent phenotype. For example, p21
waf1/cip1, or p16INK4a, and β-galactosidase enzymes are usually highly active in senescent cells; for this reason, they are frequently used as senescence markers [
6]. Moreover, lipofuscin granules, composed of carbohydrates and proteins derived from cell catabolic activity, accumulate in senescent cells and are visible as dark granules [
7]. Lastly, especially during late senescence, senescent cells produce specific inflammatory interleukins, and cytokines, with both autocrine and paracrine actions; their main function is to maintain and extend the senescent phenotype to form a barrier against the damaged cell [
8]. The ensemble of these proteins is called the Senescence-Associated Secretory Phenotype (SASP); it is one of the features most frequently associated with senescent cells, despite that fact that its components vary according to the type of senescence induced and the stage of senescence reached. Senescence establishment is a long, organized process in which every phase, from the earliest to the latest, is characterized by the production of specific mediators [
8]. In addition to these molecular changes, senescent cells are characterized by morphological changes, namely an enlarged and flattened cell shape and alterations to the nuclear membrane [
9]. It is also worth mentioning that senescent cells undergo chromatin reorganization. In dividing cells, heterochromatin is mainly compartmentalized at the nuclear periphery, while during senescence—similarly to the aging process—heterochromatin is progressively lost; however, dense foci of heterochromatin (Senescence-Associated Heterochromatin Foci—SAHF) are often visible in the nucleoplasm [
10,
11,
12]. Related to this phenomenon, epigenetic markers are also rearranged, with H3K9me2/3 and DNA methylation progressively lost globally, and especially at repetitive sequences [
10,
13]; conversely enrichment of H3K9me3 and macroH2A occurs at the SAHF [
14]. However, it has been observed that SAHF are not present in all senescent cells, as they most probably depend on cell type and stimulus [
15]. Indeed, SAHF seem to be more prominent in oncogene-induced senescence (OIS) than in replicative senescence [
11,
16]. Moreover, cells from Hutchinson–Gilford progeria syndrome patients, who undergo pathological accelerated aging, are characterized by a reduction in heterochromatin, but do not form SAHF. This suggests the existence of a two-step mechanism to first decondense chromatin and then rearrange the heterochromatin in the SAHF [
17].
Clearly, senescence induction in cancer cells can be a powerful strategy in counteracting tumor spread. In particular, the reactivation of senescence-inducing pathways—specifically in cancer cells, and not in non-cancerous immortalized cells—is an important goal to generate anti-cancer therapies that could be safer for patients. It has recently been shown that some molecules display pronounced anti-cancer activity by triggering senescence in cancer cells. For example, the natural polyphenol curcumin, derived from
Curcuma longa, has been shown to induce senescence in HCT116 cancer cells by causing cell-cycle arrest in the G2/M phase [
18,
19,
20,
21]. Herein, we investigate the mechanism/s underlying curcumin-mediated senescence, confirming its more specific action against cancer cells. We analyze several markers of senescence and look at the presence of DNA damage. Lastly, we show that induced senescent cancer cells can also be specifically cleared by fisetin and quercetin. These two flavonoids, known for their broad anti-inflammatory action, can act in cells both as senescence inducers and senolytic drugs, depending on the dose [
22]. For the latter function, flavonoids have been shown to play an important role in counteracting aging by removing senescent cells [
23,
24]. Coupling senolysis, described as the induction of apoptosis in senescent cells, with senescent inducers such as curcumin, can be a potential strategy in the development of anti-cancer therapies; this might allow the selective elimination of senescent cancer cells, without affecting the non-cancerous cells of the organism.
2. Materials and Methods
2.1. Cells and Cell Culture
HCT116 cells with the MIN phenotype (near-diploid) (kindly provided by Dr B. Vogelstein, John Hopkins University, Baltimore, MD), MCF-7, and RPE-1 hTERT cells were cultured in DMEM with 10% FBS (Corning, catalogue number 35-010-CV), 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Cells were cultured in a humidified atmosphere with 5% CO2 at 37 °C. Cells were usually divided when they reached a confluency of 90–100%. Cells were treated with 10 µM of curcumin for 24 h and then released into fresh medium. Additional treatment for 72 h with 5 or 10 μM of fisetin and quercetin was also administered. All of the used molecules (curcumin, fisetin, quercetin) (kindly provided by Prof. Riela and Dr. Massaro, University of Palermo) were dissolved in DMSO. DMSO treatment was used as a control. The DMSO percentage in both treated and control cells was lower than 0.1% of the total volume.
2.2. Cell-Cycle Analysis
Cell-cycle profiles of asynchronously growing cells were analyzed via flow cytometry. The DNA content was determined by staining cells with Propidium Iodide (PI, Merk-Sigma-Aldrich, Milan, Italy), a DNA dye that binds the DNA in a stoichiometric way. The DNA amount enables the detection of cells in each phase of the cell cycle: the G0/G1, S, and G2/M phases. Aliquots of 10
6 cells were obtained via trypsinization and collected in cold Phosphate–Buffered Solution (PBS). Cells were then harvested via centrifugation (10 min at 1000 rpm), washed with PBS, and incubated in the dark in PBS containing 20 μg/mL PI and 40 μg/mL RNase, for 30 min, at room temperature. Samples were then subjected to fluorescence-activated cell-sorting analysis using a FACSCanto flow cytometer (Becton Dickinson Biosciences). At least 20,000 cells were analyzed for each sample using the FACSDiva software, Kaluza software and FlowJo software. Discrimination of cell doublets from singlets was carried out by plotting the DNA dye (PI) channel width versus its area in a dot-plot graph as previously described [
25]. Gating was carried out by distinguishing 2n- and 4n-content cells (G1 and G2/M phase, respectively) on the base of DNA content.
2.3. SA-β-Gal Staining
Cellular senescence was evaluated using an SA-β-Gal assay. Cells were cultured in triplicate in an MW12 plate and, after medium removal and washes with PBS, fixed on the day of the analysis with 4% PFA (paraformaldehyde, pre-heated at 37 °C) for 5 min at room temperature. Cells were then washed twice with PBS for 5 min, and incubated for 24 h at 37 °C with a staining solution containing the following in 40 mM citric acid/sodium phosphate solution, pH 6.0:
X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; Promega, Milan, Italy, catalogue number V3941) 0.1%;
5 mM of potassium ferrocyanide and 5 mM of potassium ferricyanide;
150 mM Sodium chloride;
2 mM Magnesium chloride.
The staining solution was removed the next day, and cells were washed twice with distilled water; then, they were observed under a transmitted light microscope with a 20× objective. The percentage of senescent cells was evaluated on 600 cells for HCT116 and 100 cells for RPE-1 hTERT. At least two independent experiments with two technical replicas were performed.
2.4. Immunofluorescence
2.4.1. γH2AX
Cells were seeded the day before the treatment on glass coverslips, which were placed on the bottoms of wells of an MW12 plate. Cells were treated at a 40% confluence with curcumin 10 µM. On the day of the analysis, cells were washed twice with PBS and fixed with ice-cold methanol for 5 min at room temperature, or until evaporation. After two washes with PBS, cells were permeabilized with 0.01% Triton-X diluted in mQ water for 10 min at room temperature; then, they were washed again and blocked with 0.1% BSA (Bovine Serum Albumin) for 30 min at room temperature. Antibody labeling was carried out by diluting antibodies in 0.1% BSA. The primary antibody against γH2AX (Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301, Upstate®®, from mouse; catalogue number 05-636) was diluted at 1:200 and incubated overnight at 4 °C. The secondary antibody (Goat Anti-Mouse IgG H&L (Alexa Fluor®® 488), ab150113, Abcam, Cambridge, UK) was used at 1:500 and left for 1 h at room temperature. Each labeling was followed by PBS washes. DAPI (1 μg/mL) was added before observation with a Zeiss Axioskop microscope.
2.4.2. 5-Methylcytosine
For 5-Methylcytosine staining cells were grown onto coverslips, then washed with 0.1% Tween-20 in PBS and fixed for 10 min in freshly prepared 4% formaldehyde in PBS. Cells were permeabilized with 0.5% Triton X-100 in PBS for 30 min and treated with 2M HCl for 30 min at 25 °C. After extensive washing with 0.1% Tween-20 in PBS, cells were blocked for 1 h in 2% BSA in 0.1% Tween-20 in PBS; then, they were incubated with 1:500 anti-5MC antibody (5-Methylcytosine Monoclonal Antibody, Epigentek, Farmingdale, NY, USA, from mouse, clone 33D3; catalogue number A-1014) in 0.1% Tween-20 in PBS, overnight, at 4 °C; and subsequently, after washing, they were labelled with 1:500 anti-mouse Cy3-conjugated in 0.1% Tween-20 in PBS, for 1 h, at 37 °C. DNA was stained with DAPI (4′,6-diamidino-2-phenylindole) (1 µg/mL) for 5 min at room temperature, and cells were imaged with a Zeiss Axioskop microscope. The experiment was repeated three times, and at least 50 nuclei/sample were quantified for each experiment.
2.4.3. H3K9me3
For H3K9me3 staining, cells were grown onto coverslips, then washed with PBS and fixed for 10 min in freshly prepared 4% formaldehyde in PBS. Cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min. After extensive washing with PBS, cells were blocked for 1 h in 5% BSA and then incubated with 1:100 anti-H3K9me3 antibody (Anti-trimethyl Histone H3 (Lys9), clone 6F12-H4, from mouse; Merck, Milan, Italy; catalogue number 05-1242-S) in 5% BSA, for 1 h, at room temperature; after five washes, they were labelled with 1:1000 anti-mouse Cy3-conjugated in 5% BSA, for 1 h, at room temperature. DNA was stained with DAPI (1 µg/mL) for 5 min at room temperature, and cells were imaged with a Zeiss Axioskop microscope. The experiment was repeated three times, and at least 50 nuclei/sample were quantified for each experiment.
2.4.4. MPM-2
For MPM2 staining, the manufacturer’s instructions for MPM2 antibody were followed. Briefly, cells were grown onto coverslips, then washed with PBS and fixed for 10 min in freshly prepared 4% formaldehyde in PBS. Cells were permeabilized with ice-cold methanol for 5 min on ice. After extensive washing with PBS, cells were blocked for 1 h in 1% BSA at room temperature and then incubated with 1:100 of anti-phospho-Ser/Thr-Pro, MPM2 (mitotic protein monoclonal #2, from mouse; Merck, Milan, Italy, catalogue number 05-368) in 1% BSA in PBS, overnight, at 4 °C. Subsequently, after washing, cells were labeled with 1:1000 anti-mouse Cy3-conjugated (Anti-mouse IgG (H + L), Cy3 conjugated secondary antibody, Jackson ImmunoResearch; catalogue number 715-225-150) in PBS, for 1.5 h, at 37 °C. DNA was stained with DAPI (1 µg/mL) for 5 min at room temperature, and cells were imaged with a Zeiss Axioskop microscope. The experiment was repeated three times, and at least 100 nuclei/sample were quantified for each experiment.
2.5. Image Analysis
5-Methylcytosine and H3K9me3 signals were quantified by using a FIJI-based automated system. A mask of the nuclei was obtained by thresholding the DAPI channel, and individual nuclei were detected using the Analyze Particles function. Five 15 × 15-pixel circles were drawn outside the nuclei (marked by DAPI staining) and the means of the integrated signal was calculated (background). The integrated signal intensity of each nucleus was then calculated by subtracting the background as previously performed in [
26].
MPM2 staining was analyzed in FIJI as follows: cells were manually counted by using the DAPI channel. All of the images with MPM2 signal were background-subtracted, and then merged with the DAPI channel to obtain the right match between DNA and MPM2. Then, MPM2 spots were manually counted for every single nucleus.
Positivity to phospho-γH2AX staining was considered according to the presence of spots inside cells’ nuclei. Diffuse fluorescence was considered as a background signal. A total of 100 nuclei and 60 nuclei were counted for HCT116 cells and RPE-1 hTERT cells, respectively.
2.6. RT-qPCR
RNA was extracted from at least 1 × 10
6 cells using the ReliaPrep
TM RNA Cell Miniprep System (Promega, Milan, Italy). RNA was quantified using Nanodrop
TM and loaded on 1% agarose gel to verify its integrity. An amount of 1 μg of RNA was reverse-transcribed in cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Milan, Italy) with the following conditions, according to the manufacturer’s instructions:
| | Step 1 | Step 2 | Step 3 | Step 4 |
| Temperature (°C) | 25 | 37 | 85 | 4 |
| Time | 10 min | 120 min | 5 min | ∞ |
RT-qPCR was performed using Applied Biosystems 7300 Real-Time PCR System Software. Each sample was analyzed in triplicate (50 ng of cDNA/replicate), with a final volume of 25 μL for each replicate. The reaction mix was prepared as follows (the indicated volumes and concentrations are referred to as one replicate):
12.5 μL 1× Master Mix SyBR Green (Applied Biosystems, catalogue number: 4367659);
2 μM Forward- and Reverse-primer mix;
RNase-/DNase-free H2O.
Gene expression was quantified by comparing the 2
−ΔΔCt of each gene to the one of the endogenous gene GAPDH (Forward primer: 5′-CTCATGACCACAGTCCATGCC-3′; Reverse primer: 5′-CAATCCACAGTCTTCTGGGT-3′) as previously performed in [
27]. P21
waf1/cip1 sequence was targeted with the Forward primer 5′-CTGGAGACTCTCAGGGTCGA-3′ and the Reverse primer 5′-CGGATTAGGGCTTCCTCTTG-3′.
2.7. Metaphase Spread
Ploidy of the cells was analyzed using a metaphase spread assay. Cells were seeded to be at 70% of confluency on the day of the analysis on microscope slides, to avoid artifacts due to drip. On the day of the analysis, cells were treated with Colcemid (0.2 μg/mL, Merck–Sigma-Aldrich, Milan, Italy) for 2 h. After one wash in PBS, cells were incubated with 75 mM KCl solution for 20 min at 37 °C, to induce swelling. Nuclei were then fixed with ice-cold fixative (3:1 methanol:glacial acetic acid) and stained with 5% Giemsa staining solution for 8 min. Microscope slides were then rinsed with distilled H
2O before observation with 20× objective (for the search of metaphases and the calculation of mitotic index), and with the 63× and 100× for chromosome analysis and chromosome-break evaluation, as previously performed in [
27].
For each sample, at least 20 metaphases were analyzed. The experiment was repeated 4 times.
2.8. Western Blot
Cells were lysed with 1× Laemmli Buffer and the proteins denatured at 95 °C for 10 min. An amount of 20 µL of the samples were loaded together with a protein ladder marker in a 10% polyacrylamide gel. The SDS-PAGE gel was run in a Mini-PROTEAN System (Bio-Rad, Italy), with Tris-Glycin pH 8.3 running buffer (0.025 M Trizma base, 0.192 M glycin and 0.1% SDS), firstly at 50 Volts, and then at 80 Volts for the rest of the run (90 min in total). The proteins were then transferred onto a PVDF membrane, and pre-activated in methanol, for 90 min, with constant voltage (V = 100), on ice. After blocking with 5% skimmed milk for 1 h, proteins were probed with a solution of 1:1000 anti-p21waf1 (Santa Cruz Biotechnology, catalogue number sc-6246); 1:500 anti-γH2AX (Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301, Upstate®®, from mouse; catalogue number 05-636); 1:2000 anti-p53 (Santa Cruz Biotechnology); 1:10,000 anti-β-Tubulin (Sigma-Aldrich, catalogue number T4026); and 1:5000 anti-actin (Thermo Fisher Scientific, Milan, Italy), in 0.5% skimmed milk, and incubated while agitating for 1 h, at room temperature. Incubation with anti-Mouse-HRP secondary antibody (Thermo Fisher Scientific, Milan, Italy) followed, for 1 h at room temperature. Protein bands were revealed using Chemidoc XRS+ 60 (Bio-Rad, Italy) following SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Milan, Italy) incubation. Bands were quantified using ImageJ software as follows: A box was drawn around the bands, including all of the intensity of the bands, with a minimal amount of surrounding background. In the immediate surrounding of each band, an additional region of interest was drawn to designate the background region. The signal within the boxes was quantified. The value of background box was then subtracted from all of the bands’ boxes. To perform normalization, the control sample was used as the reference. The ratio of the signals of the housekeeping protein in each lane, using the reference lane as the numerator, yielded the difference in sample load between the reference and the other samples. This normalization factor was then multiplied by the protein signal for each sample. The mean of at least three replicates was represented in the graphs.
2.9. SAHF Staining
Cells were stained as previously reported by Aird and Zhang [
28]. Briefly, cells were seeded onto glass coverslips for the duration of the treatment, then washed with PBS and fixed for 10 min in freshly prepared 4% formaldehyde in PBS. After extensive washing with PBS, cells were permeabilized by incubating coverslips in 0.2% Triton X-100 in PBS for 5 min at room temperature and then blocked for 5 min in 3% BSA at room temperature. DNA was stained with DAPI (0.15 µg/mL) in 3% BSA for 3 min at room temperature. Cells were extensively washed with PBS, then imaged with a Zeiss Axioskop microscope. The experiment was repeated twice, and at least 100 nuclei/sample were observed.
2.10. AO/EB Assay
HCT116 cells were seeded in a 60 mm Petri dish to reach the 40% of confluency on the day of curcumin treatment. At the end of senolytics treatment, cells were trypsinized, and then collected in cold PBS. Culture media were collected too. Then, cells were centrifuged for 5 min at 1000 rpm; the pellet was resuspended, washed once with PBS, and centrifuged for 5 min at 1000 rpm. Cells were then resuspended in 270 µL of a mix containing AO/EB double-staining solution (1×) and PBS, and the mix was poured on glass coverslips for microscopic observation. A total of 100 cells were counted to quantify each condition.
2.11. Clonogenic Assay
1000 HCT116 cells were seeded on 6-well plates, and at the end of treatments with curcumin and senolytics, colonies were fixed for 10 min in methanol. After washing with PBS, cells were stained for 10 min using a crystal violet staining solution (1% crystal violet, 20% EtOH). After washing with PBS, cells were left to dry upside-down, and then images were taken with a scanner.
2.12. Trypan Blue Staining
Cell samples were diluted 1:1 in 0.4% Trypan blue solution. Then, cells were counted under a microscope in three 1 × 1 mm squares of a Burker chamber, to determine the average number of cells per square (total cell number and Trypan Blue positive cell number); this was multiplied by 104 to obtain the number of cells per milliliter, and corrected for the dilution.
4. Discussion
Cell senescence can be induced by a plethora of stressors and stimuli, including telomere shortening (replicative senescence), oncogene activation (oncogene-induced senescence, OIS), mitochondrial stress, cell fusions, oxidative stress, and inflammatory cytokines [
32]. Most of these lead to DNA damage, which, in turn, triggers permanent cell-cycle arrest (senescence). In the most recent years, it has been also demonstrated that mitotic stress can promote cell-cycle arrest of OIS [
30]. Extended mitotic arrest could result in either abnormal cell division with aneuploidy generation after the arrest, in mitotic catastrophe and cell death, or in mitotic slippage whereby cells bypass cell division, returning to interphase with tetraploid DNA content (reviewed in [
33]). Then, the mitosis-bypassing cells undergo cell death or cell senescence after slippage [
31,
34,
35,
36]. If the cells survive the mitotic slippage, endoreduplication usually occurs, and cells become giant and accumulate DNA damage [
31,
37]. All of this triggers the transition to cell senescence. Here, we show that a similar fate characterizes cancer cells treated with curcumin. Curcumin shows an elevated degree of pleiotropy, which is responsible for the heterogeneous responses observed following its administration. Furthermore, curcumin triggers the downregulation of many genes involved in the inflammatory response, displaying anti-inflammatory activity, as well as the downregulation of anti-apoptotic proteins [
20,
21]. Lastly, it has been related to the HAT p300 inhibition and to a reduction in CDK1 transcription, which probably underlies the G2/M arrest observed in cancer cells [
38].
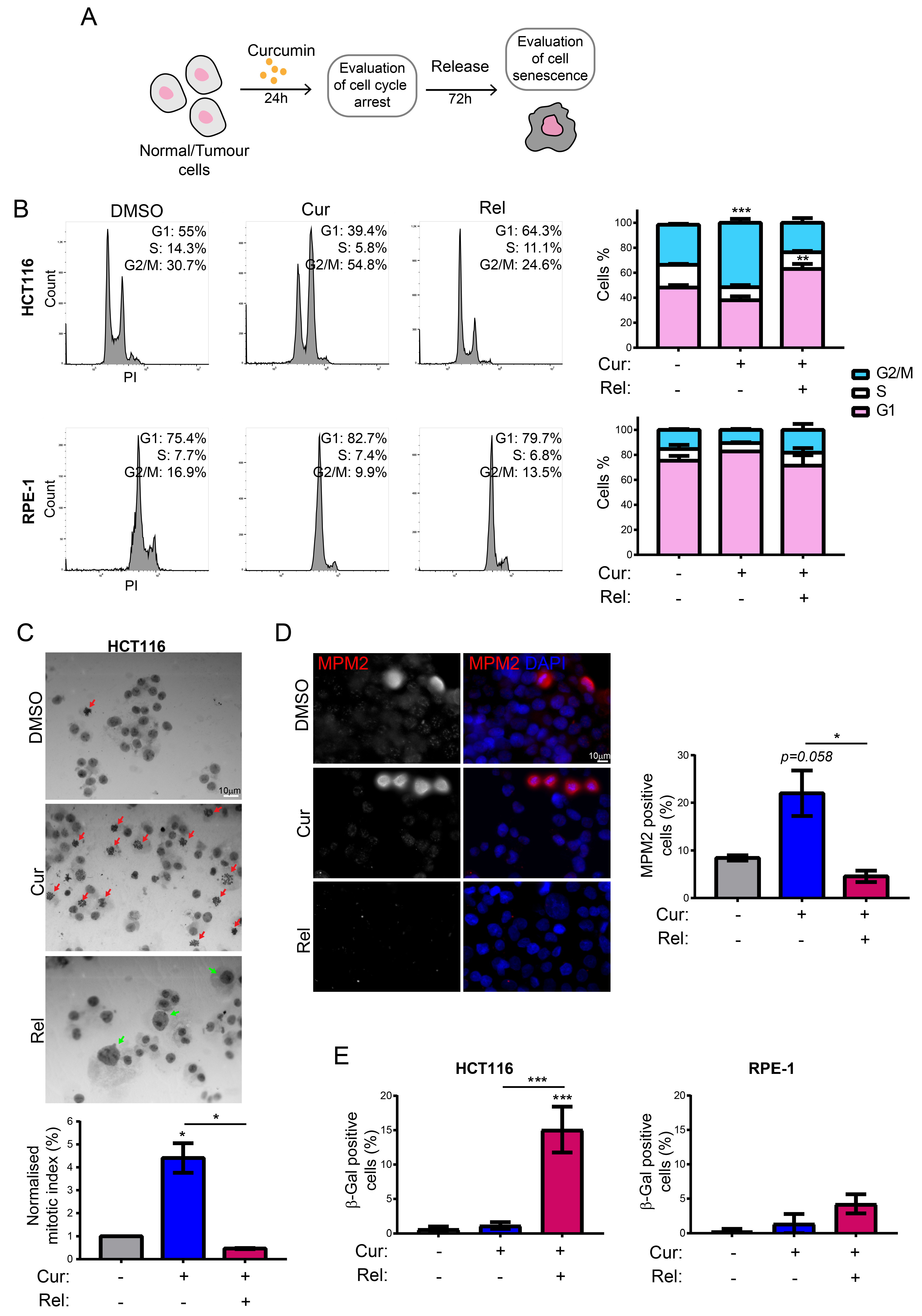
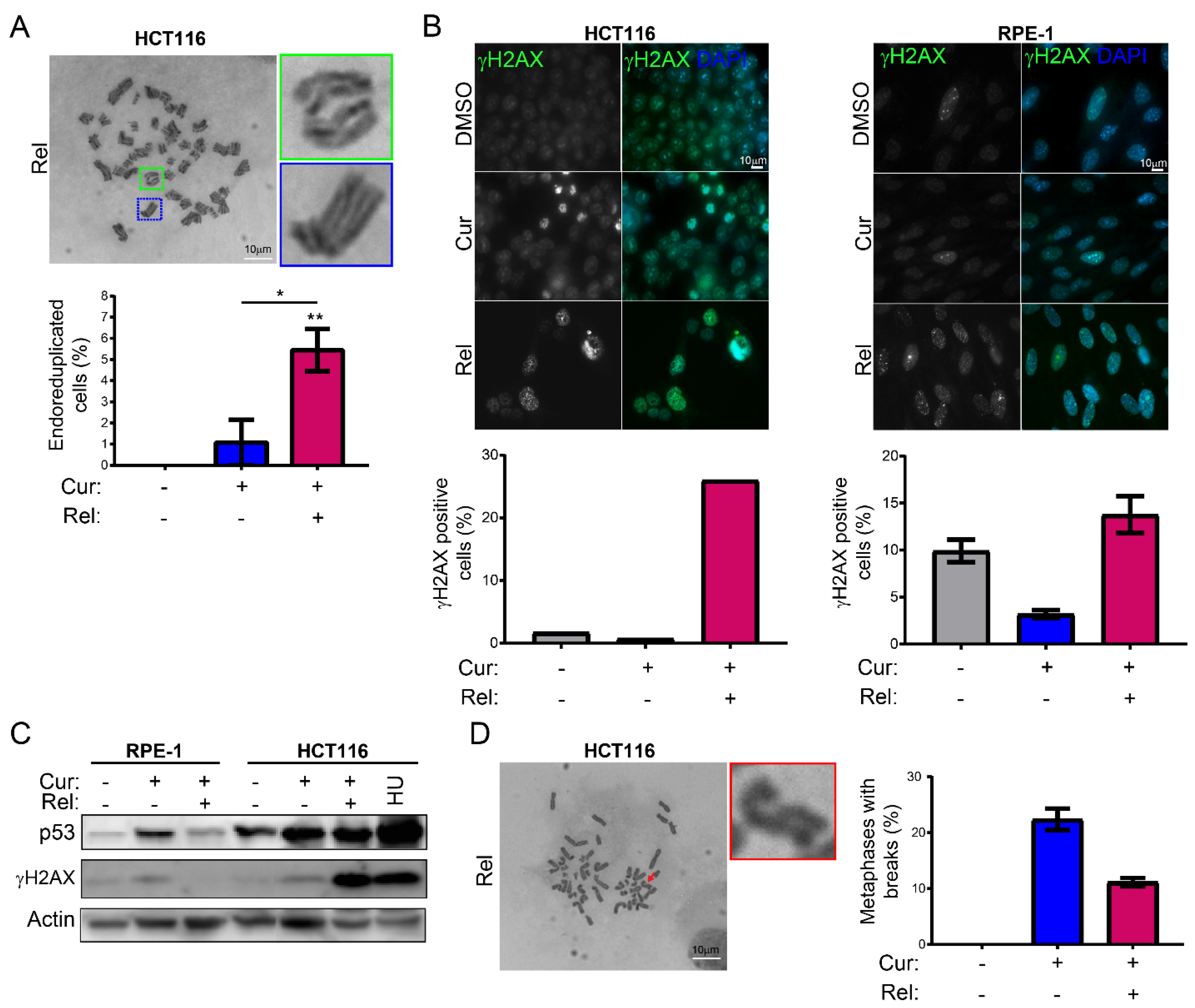
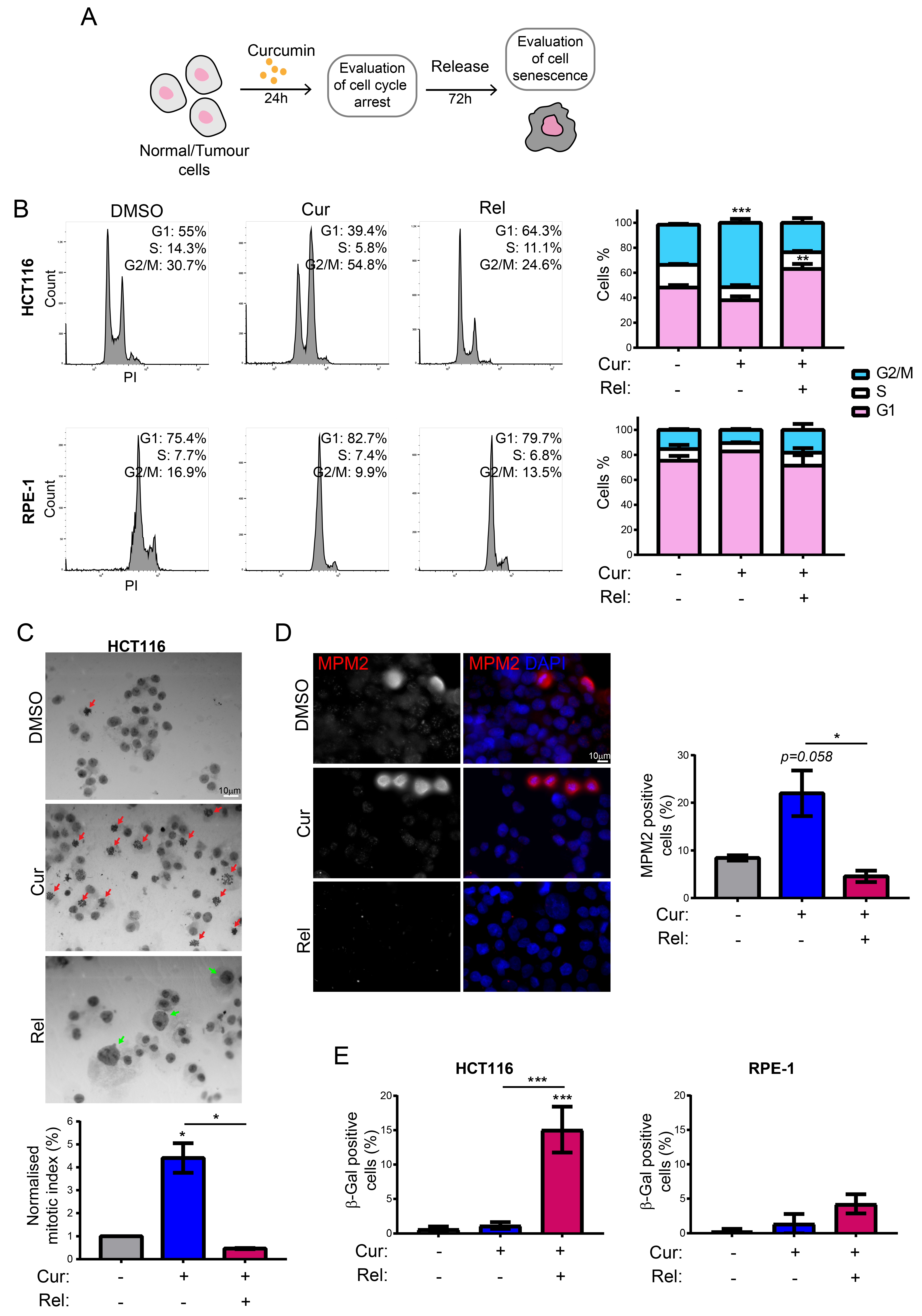
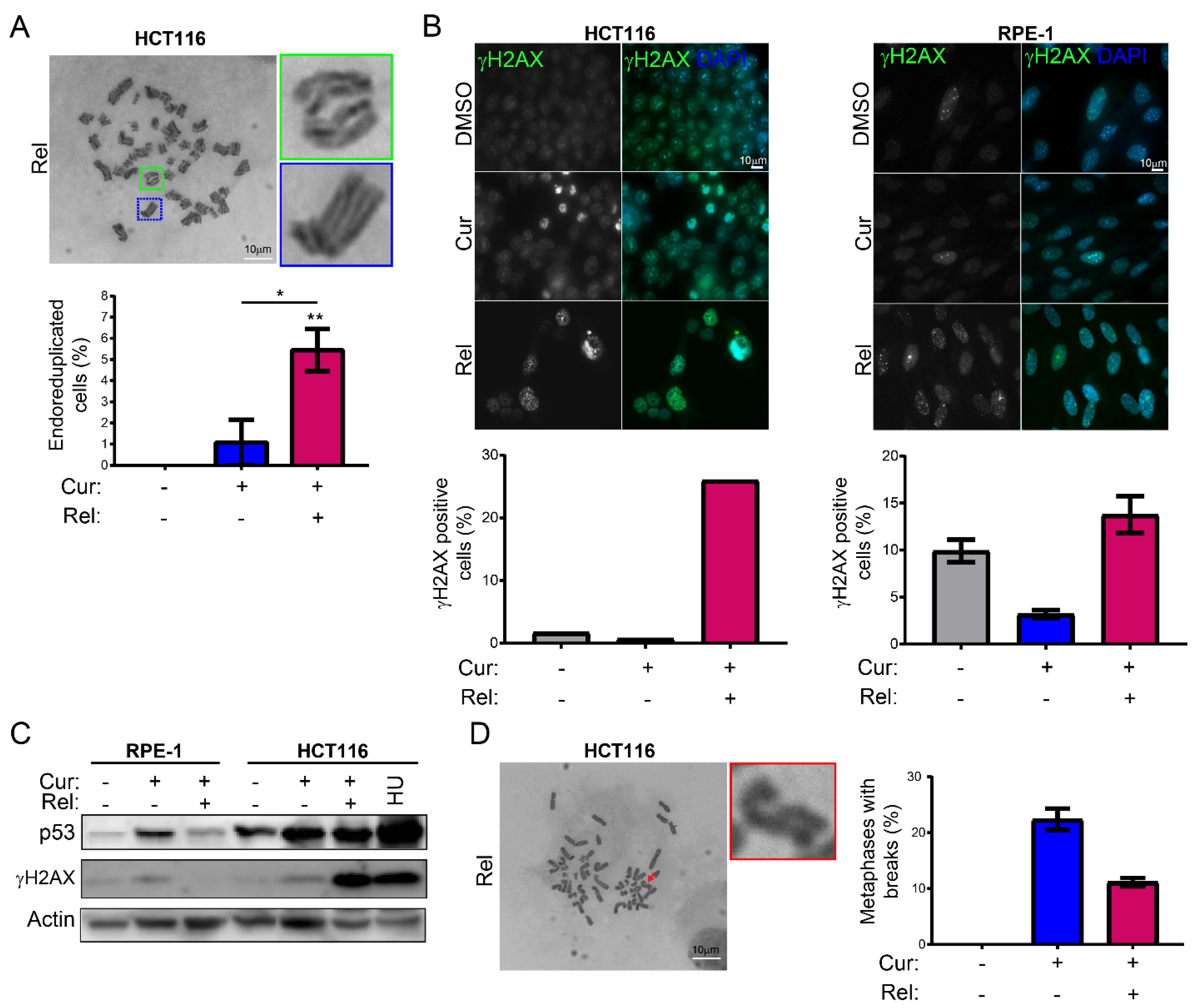
We observed that curcumin first induces selective cell-cycle arrest in mitosis only in cancer cells, and not in non-cancerous immortalized cells. When cancer cells are then released from curcumin, they can escape mitotic arrest, undergoing mitotic slippage and, partly, endoreduplication. In fact, in cancer cells, we observed the appearance of endoreduplicated chromosomes (
Figure 2A) and big nuclei suggestive of a hyperdiploid content. We also noticed that the escaped cells accumulate chromosome breaks and DNA damage (γH2AX foci and protein increase) (
Figure 2B–D). Curiously, curcumin treatment for 24 h induced an increase in the tumor suppressor p53 in both HCT116 and RPE-1 cells, suggesting the activation of the DNA damage checkpoint. However, while RPE-1 cells fully recovered during curcumin release (a reduction to normal levels of p53 and γH2AX proteins,
Figure 2C), HCT116 cells underwent a further increase in γH2AX and p53, suggesting that in this cancer-cell line, curcumin insult cannot be fixed. All of this probably induces the observed senescence in cancer cells. Senescence induction, as previously reported [
18], was, indeed, supported by SA-β-Gal activity, which highly increased, specifically in curcumin-released cancer cells, compared to the untreated condition (
Figure 1E). Senescence induction was also suggested by the larger and flatter morphology observed in the cells released from the treatment. The result is particularly striking when compared to the non-cancerous immortalized cells, which display no significant increase in SA-β-Gal activity upon release from curcumin treatment (
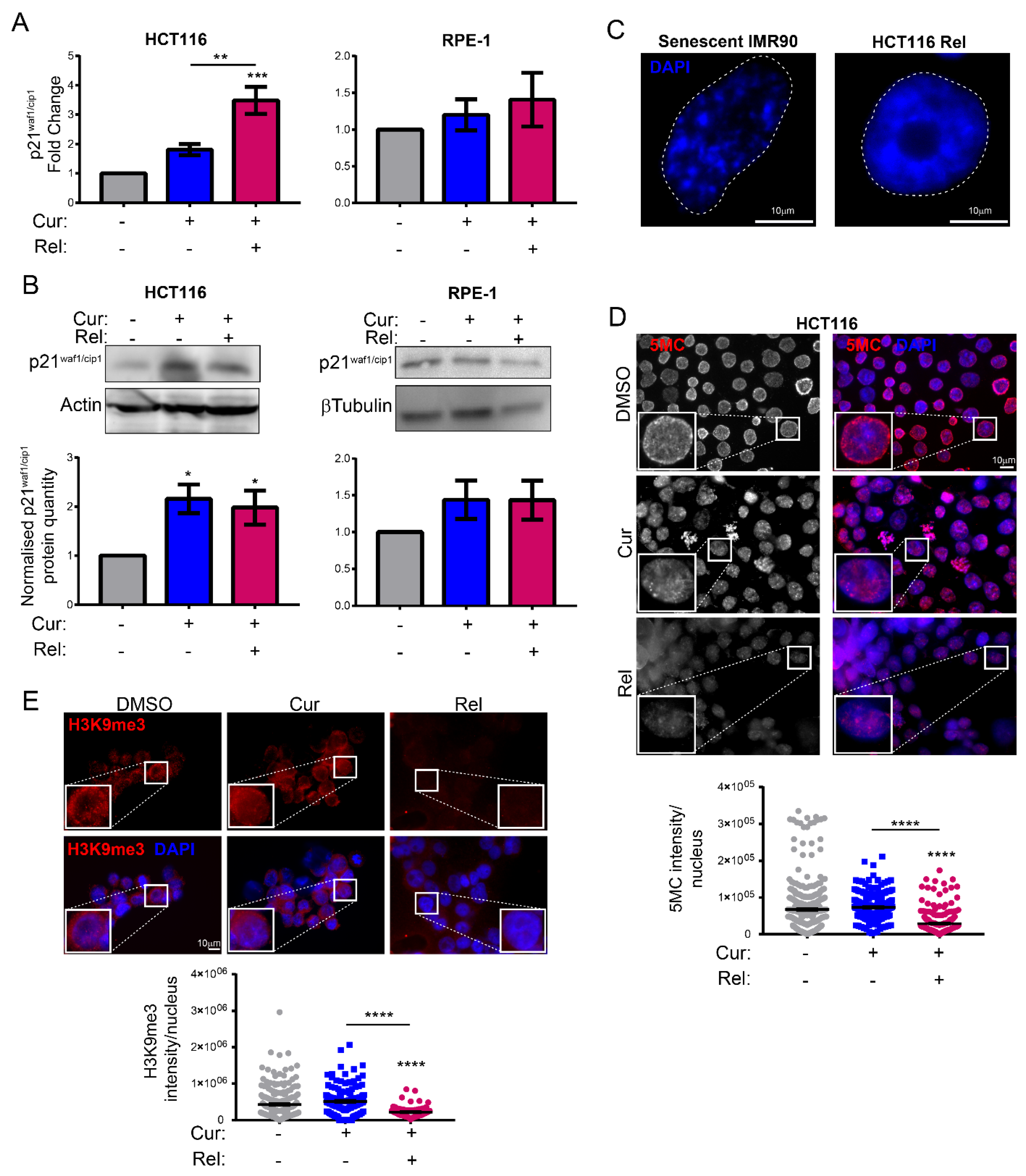
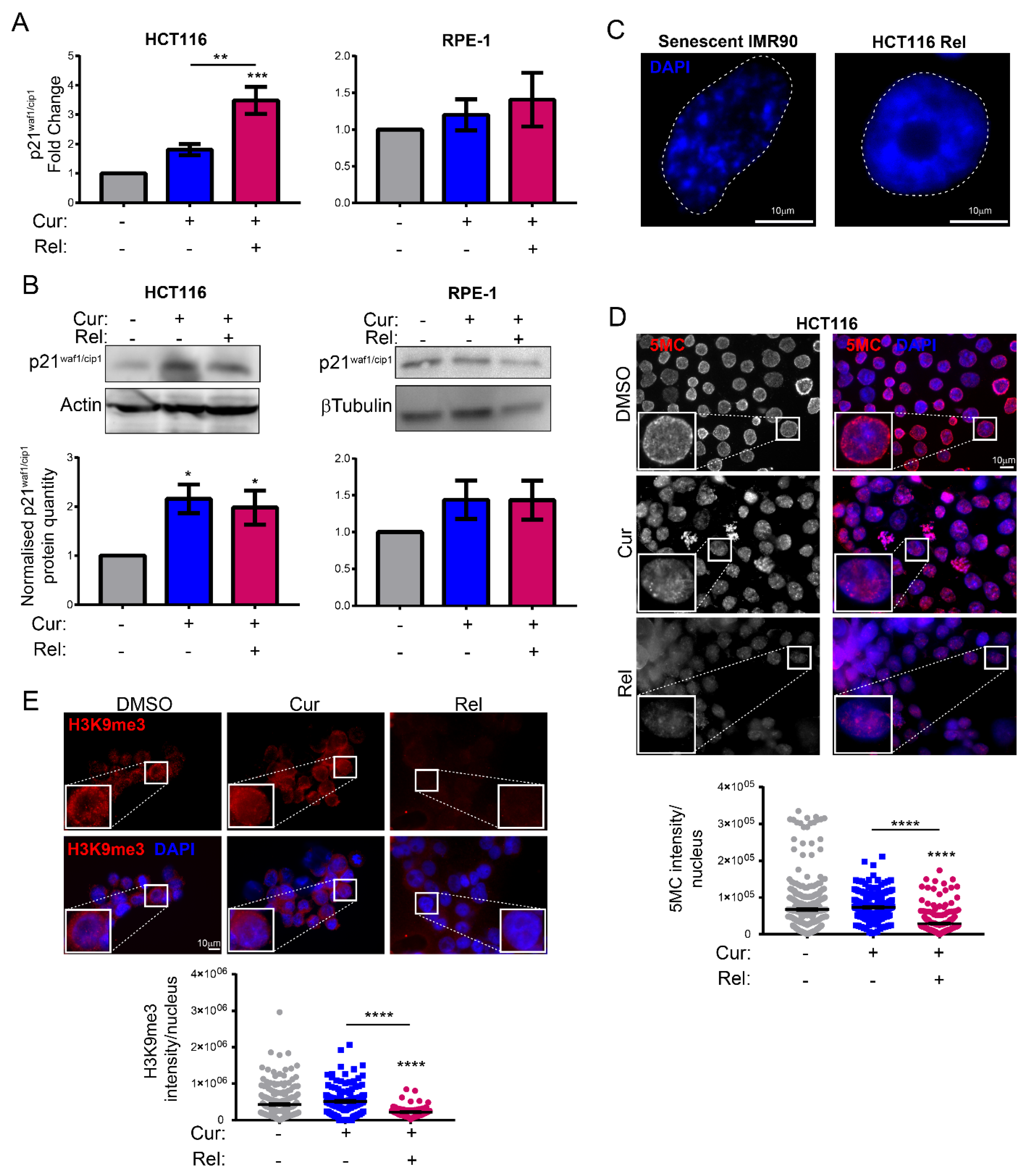
Figure 1E). This finding suggests the selectivity of curcumin action towards cancer cells. We show that curcumin-induced senescence depends on p21
waf1/cip1 activation, which further confirmed the upstream activation of p53. We also demonstrate that curcumin-induced senescent cancer cells undergo global heterochromatin loss, assessed by the nuclear reduction in the heterochromatin markers DNA methylation (5MC) and H3K9me3. Moreover, the localization of the staining of these markers in the nuclei changes, moving away from the nuclear periphery towards the nucleoplasm. This matches with the epigenetic rearrangements that cells experience during senescence [
13]. However, we have not observed SAHF in curcumin-induced senescent cancer cells, which is similar to what was observed in other senescence contexts, and which could reinforce the hypothesis that SAHF formation is a two-step mechanism [
17]. We tested senescence markers of the early phases of senescence establishment that are not compromised by the side effects of flavonoids. On the other hand, SASP is an important feature of senescent cells, at least in overt senescent cells. However, in this case, SASP might not be involved, because of the well-known curcumin function in the de-regulation of genes related to the inflammatory response. It has been shown that curcumin strongly reduces NF-κB and IL-1α expression [
39,
40], which are the main protein factors responsible for SASP initiation and maintenance. However, SASP occurrence could be tested to evaluate its cause–effect relationship with a persistent and transient DNA damage response [
41,
42].
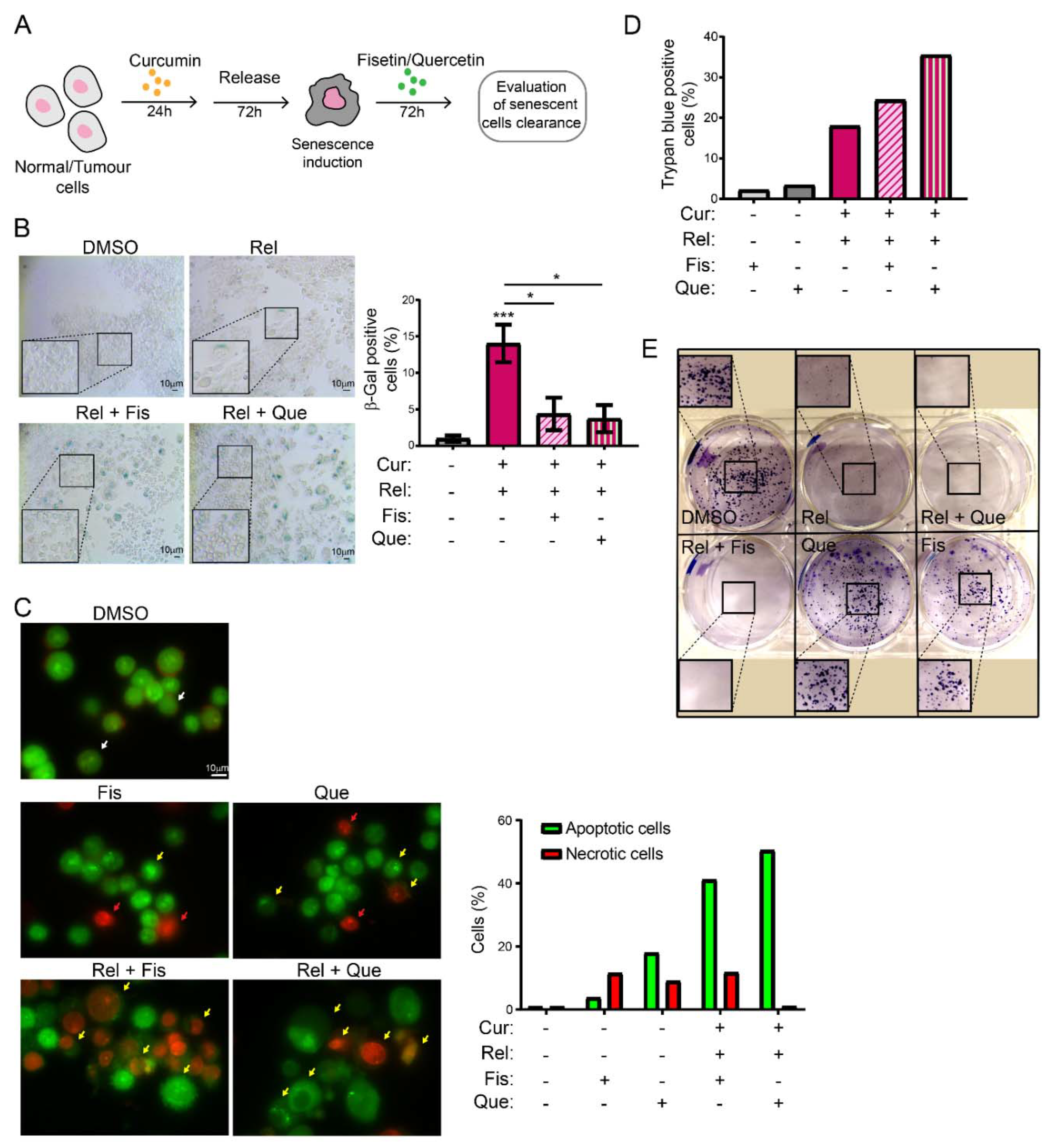
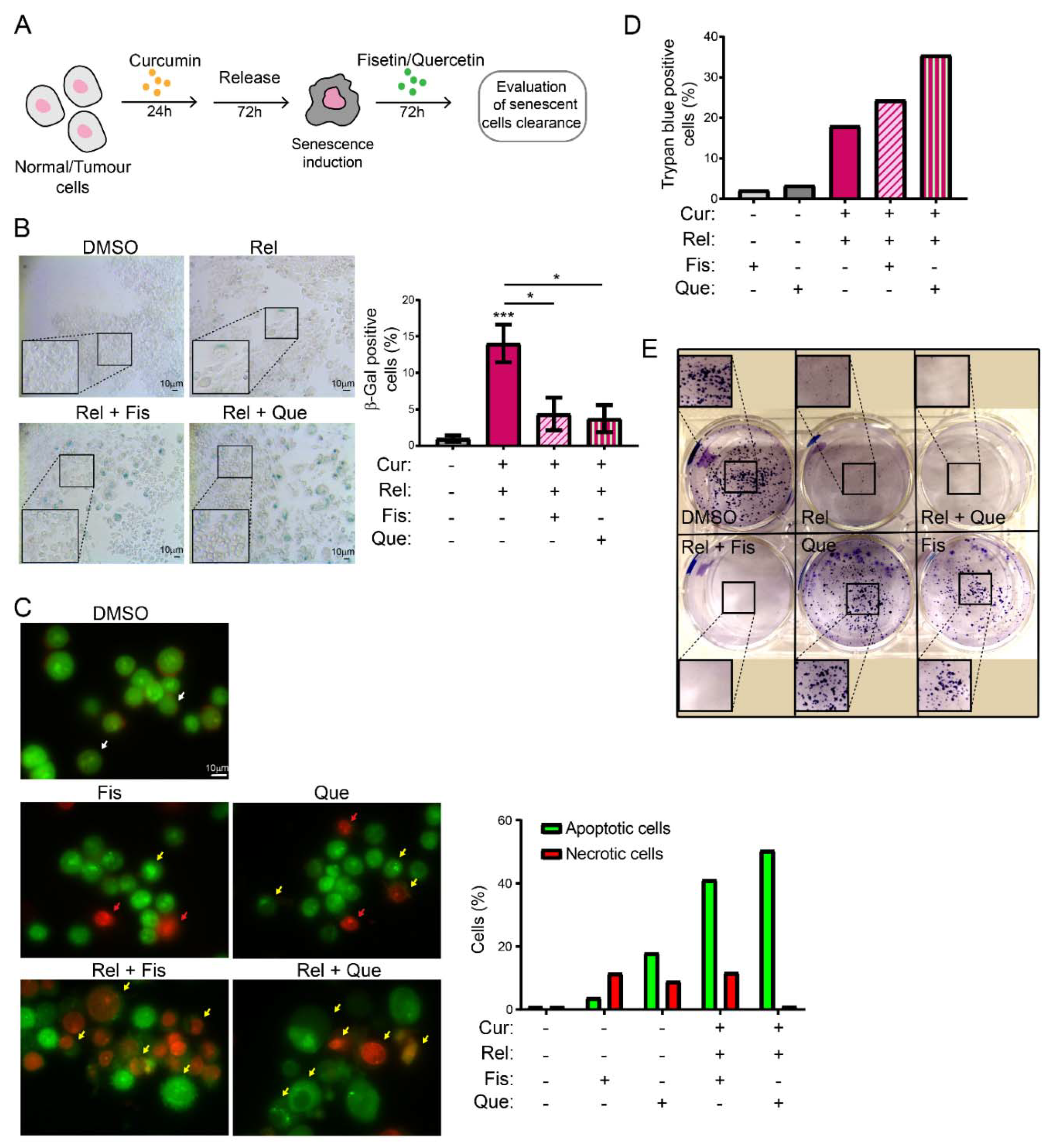
Selectively inducing senescence only in cancer cells and not in non-cancerous immortalized cells could be, by itself, an interesting strategy to block cancer cells’ proliferation in patients. However, the persistence of a therapy that induces senescence could be detrimental in the long term. Thus, the research of strategies aimed to eliminate therapy-induced senescent cells has recently acquired interest. This would minimize both the collateral effect of long-term treatment with senescence-inducing activity, and the possibility of tumor resumption. There are mainly two lines of research in this context: targeting senescent cancer cells with senolytic drugs or with senolysis mediated by the immune response (reviewed in [
43]). Thus, we explored the possibility of combining curcumin treatment with the administration of other natural compounds with senolytic activity, in an attempt to clear curcumin-induced senescent cells. The obtained results suggest that fisetin and quercetin reduce the number of curcumin-induced senescent cells by inducing an apoptotic response, which is coherent with the current knowledge of senolytics’ action. In fact, AO/EB staining revealed cells whose morphology, following curcumin and senolytic treatments, was dramatically altered, with membrane blebs and chromatin fragmentations which are typical of apoptosis. The bright green and yellow staining of cells treated with curcumin and senolytics, as well as the red staining of nuclei and chromatin, reinforces the assumption that apoptosis was induced, since these features are suggestive of early and late apoptosis, respectively. Ethidium bromide can only stain DNA red if the plasma membrane integrity is lost, a phenomenon that occurs only during apoptosis or necrosis. However, this last outcome can easily be excluded, since the green staining (which is typical of viable or early apoptotic cells), membrane blebs and apoptotic vesicles are also detectable. In parallel, the number of non-cancerous, not-senescent, proliferating cells increases following treatment with senolytics, meaning that their action is specific and does not affect non-cancerous immortalized cells. On the contrary, in cancer cells, viability assays displayed a cytotoxic response, as well as an impairment of cell proliferation, following curcumin + senolytics treatments. This outcome, combined with the general anti-inflammatory action of flavonoids, represents a potential strategy which can be safer for patients, given that a chronic inflammatory state has often been related to enhanced tumorigenesis. Our observations, thus, pave the way to a promising therapeutical strategy against cancer, based on natural compounds.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}