
Analysis of Complementary Sex-Determiner (csd) Allele Diversity in Different Honeybee Subspecies from Italy Based on NGS Data

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction, Library Preparation, Sequence Processing and Alignment
2.3. De Novo Assembly and Analysis of Sample-Specific HVR Allele Consensus Sequences
3. Results
3.1. Csd HVR Allele Reconstruction and Classification
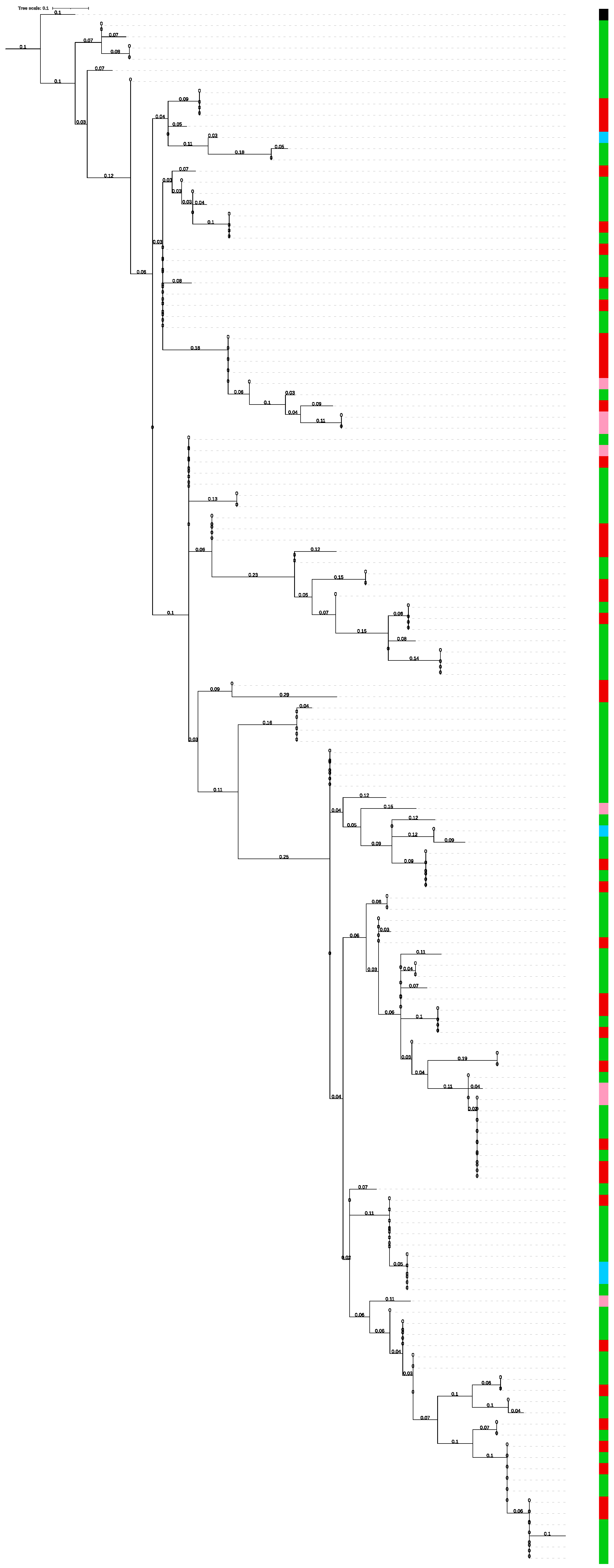
3.2. Phylogenetic Tree
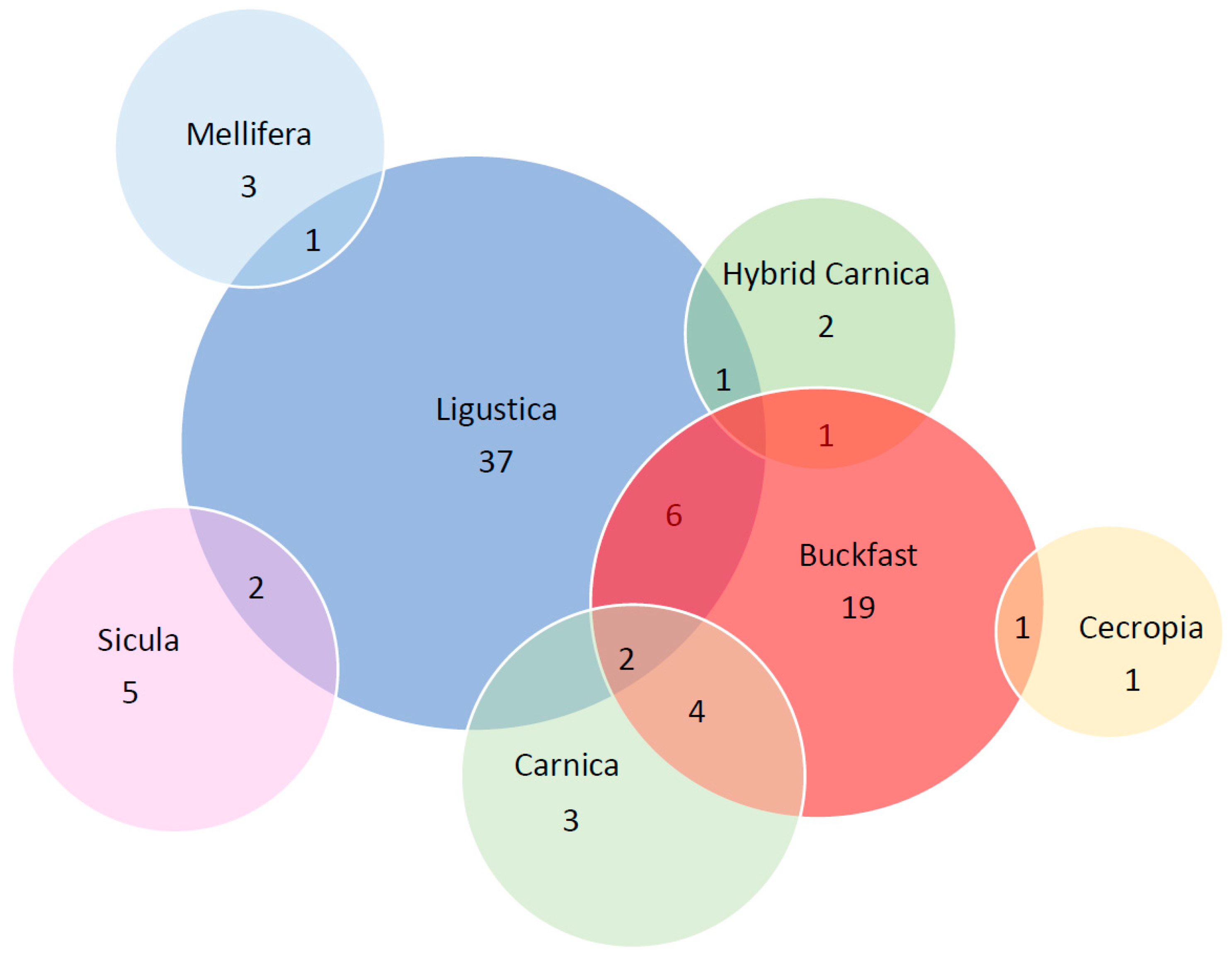
3.3. Frequency Analysis
- Among the 68 sequences belonging to 34 A. m. ligustica specimens, we identified 49 different alleles. Among these, alleles 1, 11 and 32 were the most frequent in A. m. ligustica.
- Among the 42 sequences belonging to 21 Buckfast individuals, we identified 33 different alleles. Allele 9 was the most frequent, with a frequency of 9.52%.
- Among the eight sequences belonging to four A. m. sicula, seven different alleles were identified; allele 30 was the most frequent, with a frequency of 25%,
- Among the 10 sequences belonging to five A. m. carnica, we identified nine different alleles with a frequency of 20%; allele 25 was the most frequent.
- The remaining three subspecies (hybrid Carnica, A. m. mellifera, A. m. cecropia) had a limited number of reconstructed csd HVR sequences (4, 4 and 2, respectively), each bearing a different allele.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dzierzon, J. Neue verbesserte Bienen-Zucht des Pfarrers Dzierzon zu Carlsmarkt in Schlesien; Ernst’schen: Quedlinburg, Germany, 1861. [Google Scholar]
- Beye, M.; Hasselmann, M.; Fondrk, M.; Page, R.E.; Omholt, S.W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 2003, 114, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Mackensen, O. Viability and sex determination in the honeybee (Apis mellifera L.). Genetics 1951, 36, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Woyke, J. What happens to diploid drone larvae in a honeybee colony. J. Apic. Res. 1963, 2, 73–75. [Google Scholar] [CrossRef]
- Hasselmann, M.; Gempe, T.; Schiott, M.; Nunes-Silva, C.G.; Otte, M.; Beye, M. Evidence for the evolutionary nascence of a novel sex determination pathway in honeybees. Nature 2008, 454, 519–522. [Google Scholar] [CrossRef]
- Gempe, T.; Hasselmann, M.; Schiøtt, M.; Hause, G.; Otte, M.; Beye, M. Sex determination in honeybees: Two separate mechanisms induce and maintain the female pathway. PLoS Biol. 2009, 7, e1000222. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, M.; Beye, M. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. USA 2004, 101, 4888–4893. [Google Scholar] [CrossRef] [Green Version]
- Beye, M.; Seelmann, C.; Gempe, T.; Hasselmann, M.; Vekemans, X.; Fondrk, M.K.; Page, R.E., Jr. Gradual molecular evolution of a sex determination switch through incomplete penetrance of femaleness. Curr. Biol. 2013, 23, 2559–2564. [Google Scholar] [CrossRef] [Green Version]
- Lechner, S.; Ferretti, L.; Schöning, C.; Kinuthia, W.; Willemsen, D.; Hasselmann, M. Nucleotide variability at its limit? Insights into the number and evolutionary dynamics of the sex-determining specificities of the honey bee Apis mellifera. Mol. Biol. Evol. 2014, 31, 272–287. [Google Scholar]
- Kaskinova, M.D.; Nikolenko, A.G. Csd gene of honeybee: Genetic structure, functioning, and evolution. Russ. J. Genet. 2017, 53, 297–301. [Google Scholar] [CrossRef]
- Laidlaw, H.H.; Gomes, F.P.; Kerr, W.E. Estimation of the number of lethal alleles in a panmitic population of Apis mellifera L. Genetics 1956, 41, 179–188. [Google Scholar] [CrossRef]
- Adams, J.; Rothman, E.D.; Kerr, W.E.; Paulino, Z.L. Estimation of the number of sex alleles and queen matings from diploid male frequencies in a population of Apis mellifera. Genetics 1977, 86, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Huang, Z.Y.; Green, D.R.; Smith, D.R.; Zhang, J. Evolution of the complementary sex-determination gene of honey bees: Balancing selection and trans-species polymorphisms. Genome Res. 2006, 16, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselmann, M.; Vekemans, X.; Pflugfelder, J.; Koeniger, N.; Koeniger, G.; Tingek, S.; Beye, M. Evidence for convergent nucleotide evolution and high allelic turnover rates at the complementary sex-determiner gene of western and asian honeybees. Mol. Biol. Evol. 2008, 25, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Hyink, O.; Laas, F.; Dearden, P.K. Genetic tests for alleles of complementary sex-determiner to support honeybee breeding programmes. Apidologie 2013, 44, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Kaskinova, M.D.; Gataullina, A.R.; Saltykova, E.S.; Gaifullinaa, L.R.; Poskryakov, A.V.; Nikolenko, A.G. Polymorphism of the Hypervariable Region of the csd gene in the Apis mellifera L. population in Southern Ural. Russian J. Gen. 2019, 55, 267–270. [Google Scholar]
- Kolics, É.; Parrag, T.; Házi, F.; Szepesi, K.; Heltai, B.; Mátyás, K.; Kutasy, B.; Virág, E.; Taller, J.; Orbán, L.; et al. An alternative, high throughput method to identify csd alleles of the honey bee. Insects 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Taurisano, V.; Schiavo, G.; Bolner, M.; Fontanesi, L. Application of Next Generation semiconductor-based sequencing for the identification of Apis mellifera complementary sex-determiner (csd) alleles from honey DNA. Insects 2021, 12, 868. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Wu, X.; Yan, W.; Zeng, Z. Polymorphism analysis of csd gene in six Apis mellifera subspecies. Mol. Biol. Rep. 2012, 39, 3067–3071. [Google Scholar] [CrossRef]
- Zareba, J.; Blazej, P.; Laszkiewicz, A.; Sniezewski, L.; Majkowski, M.; Janik, S.; Cebrat, M. Uneven distribution of complementary sex-determiner (csd) alleles in Apis mellifera population. Sci. Rep. 2017, 7, 2317. [Google Scholar] [CrossRef]
- Bilodeau, L.; Elsik, C. A scientific note defining allelic nomenclature standards for the highly diverse complementary sex-determiner (csd) locus in honey bees. Apidologie 2021, 52, 749–754. [Google Scholar] [CrossRef]
- Wallberg, A.; Han, F.; Wellhagen, G.; Dahle, B.; Kawata, M.; Haddad, N.; Simões, Z.L.P.; Allsopp, M.H.; Kandemir, I.; De La Rúa, P.; et al. Worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis Mellifera. Nat. Genet. 2014, 46, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridi, R.; Tabet Aoul, N.; Catays, G.; Basso, B.; Bienefeld, K.; Gregorc, A.; Vignal, A.; Canale-Tabet, K. Genetic diversity and population genetic structure analysis of Apis mellifera subspecies in Algeria and Europe based on complementary sex-determiner (csd) gene. Apidologie 2022, 53, 4. [Google Scholar] [CrossRef]
- Minozzi, G.; Lazzari, B.; De Iorio, M.G.; Costa, C.; Carpana, E.; Crepaldi, P.; Rizzi, R.; Facchini, E.; Gandini, G.; Stella, A.; et al. Whole-Genome sequence analysis of Italian honeybees (Apis mellifera). Animals 2021, 11, 1311. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Meth. Mol. Biol. 2014, 1079, 105–116. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [Green Version]
- Franck, P.; Garnery, L.; Celebrano, G.; Solignac, M.; Cornuet, J.M. Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and Sicily (A. m. sicula). Mol. Ecol. 2000, 9, 907–921. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Allele ID | Amino Acid Sequences | L | Freq. in the Population | Freq. in the Subspecies | GenBank ID |
|---|---|---|---|---|---|
| Allele 1 | SSLSNKTIHNNNNYKYNYNNNNYNNNNYNNNYNNNCKKLYYNINYIEQI | 49 | 2.17% | Ligustica: 4.41% | |
| Allele 2 | SSLSNNYNYNNYNNNYKPLYYNINYIEQI | 29 | 2.17% | Ligustica: 2.94% | AAS86659.1, AAS86660.1, AAS86661.1, AAS86663.1, ABD14105.1, ABD14106.1, ABD14107.1, ABD14108.1, AGA84527.1 |
| Buckfast: 2.38% | |||||
| Allele 3 | SSLSNNYNSNSYNNYNNNYKKLQYYNIINIEQI | 33 | 1.45% | Ligustica: 2.94% | CCF23480.1, QEN96041.1 |
| Allele 4 | SSLSNNTIHNNNYKYNYNNNYNNYNNYKKLYYNINYIEQI | 40 | 1.45% | Ligustica: 1.47% | |
| Buckfast: 2.38% | |||||
| Allele 5 | SSLSNKTIHNNNNYKYNYNNNYNNNNNYSKKLYYNINYIEQI | 42 | 2.17% | Ligustica: 4.41% | AQZ41186.1, AQZ41196.1, AQZ41212.1, QEN96050.1 |
| Allele 6 | SSLSNNYNYSNYNNYNNYNNNYNNYKKLYYNINYIEQI | 38 | 1.45% | Ligustica: 2.94% | CCF23508.1 |
| Allele 7 | SSLSKNTIHNNNYKYNYNNNNNYNNNYKKLQYYNINYIEQI | 41 | 2.90% | Ligustica: 2.94% | CCF23518.1 |
| Buckfast: 4.76% | |||||
| Allele 8 | SSLSNSCNYSNNYYNKKLYYNIINIEQI | 28 | 2.17% | Ligustica: 2.94% | CCF23469.1 |
| Buckfast: 2.38% | |||||
| Allele 9 | SSLSNKTIHNNNNYKYNYNNKYNYNNNNYNKKLYYKNYIINIEQI | 45 | 3.62% | Ligustica: 1.47% | ABD14097.1, QEN96103.1 |
| Buckfast: 9.52% | |||||
| Allele 10 | SSLSNNYNYSNYNNYNNNYNNYKKLYYNINYIEQI | 35 | 1.45% | Ligustica: 2.94% | ABV56215.1 |
| Allele 11 | SSLSNNYISNISNYNNNNNSKKLYYNINYIEQI | 33 | 2.17% | Ligustica: 4.41% | AGZ61876.1, CCF23481.1, CCF23482.1 |
| Allele 12 | SSLSKNTIHNNNYKYNYNNNNYNNSKKLYYNINYIEQI | 38 | 1.45% | Ligustica: 2.94% | CCF23507.1 |
| Ligustica: 1.47% | |||||
| Allele 13 | SSLSNKTIHNNNNYKYNYNNNNYKNYNNYKKLYYNINYIEQI | 42 | 2.17% | Carnica: 10.00% | AQZ41220.1 |
| Buckfast: 2.38% | |||||
| Allele 14 | SSLSNKTIHNNNNYNNYKKLYYNIINIEQI | 30 | 1.45% | Ligustica: 2.94% | |
| Allele 15 | SSLSNNTIHNNNYKYNNYNNYNKKLYYNIINIEQI | 35 | 1.45% | Ligustica: 2.94% | CCF23492.1 |
| Carnica: 10.00% | |||||
| Allele 16 | SSLSNKTIHNNNNYNNNNYNNYKKLYYNINYIEQI | 35 | 2.17% | Buckfast: 2.38% | |
| Ligustica: 1.47% | |||||
| Allele 17 | SSLSNNTIHNNNNYNKKLYYNIINIEQI | 28 | 1.45% | Ligustica: 1.47% | AGZ61866.1, AGZ61871.1, AGZ61872.1, AGZ61874.1, CCF23470.1 |
| Buckfast: 2.38% | |||||
| Allele 18 | SSLSNKTIHNNNNYKYNYNNNCKKLYYNINYIEQI | 35 | 1.45% | Hyb. Carn: 25.00% | ABD14096.1, QEN96024.1, QEN96028.1 |
| Ligustica: 1.47% | |||||
| Allele 19 | SSLSNNYKYSNYNNYNNNYNNNYNNNYNNNYKKLYKNYIINIEQI | 45 | 2.90% | Hyb. Carn: 25.00% | ABD14145.1, ABD14146.1, ADJ57940.1 |
| Buckfast: 7.14% | |||||
| Allele 20 | SSLSNNYNSNNYNKYNYNNSKKLYYNINYIEQI | 33 | 2.17% | Buckfast: 7.14% | CCF23483.1 |
| Allele 21 | SSLSNKTIHNNNNYNNNNYNNYKKLYYNIINIEQI | 35 | 1.45% | Carnica: 10.00% | AEI99777.1 |
| Buckfast: 2.38% | |||||
| Allele 22 | SSLSNNYKYSNYNNYNNNYNNYNNNYNNNYKKLYYNINYIEQI | 43 | 1.45% | Carnica: 10.00% | CCF23533.1 |
| Buckfast: 2.38% | |||||
| Allele 23 | SSLSNHYNYNNNKYNNYNNDYKKLYYNINYIEQI | 34 | 1.45% | Ligustica: 2.94% | ADJ57943.1, AQZ41194.1, AQZ41195.1 |
| Allele 24 | SSLSNKTIHNNNNYKYNYKNYNNSKKLYYNVINIEQI | 37 | 1.45% | Buckfast: 2.38% | AEI99780.1, AEI99781.1, AEI99790.1 |
| Cecropia: 50.00% | |||||
| Allele 25 | SSLSNKTIHNNNNYNNYKKLYYNINYIEQI | 30 | 1.45% | Carnica: 20.00% | AQZ41206.1, AQZ41207.1, AQZ41208.1, CCF23474.1 |
| Allele 26 | SSLSNKTIHNNNKYNYNKYNYNNNNYNNYKKLYYNINYIEQI | 42 | 1.45% | Carnica: 10.00% | CCF23526.1 |
| Buckfast: 2.38% | |||||
| Allele 27 | SSLSNNYNYNNNNYNNYNNNYNNNYNKKLYYNIINIEQI | 39 | 2.17% | Carnica: 10.00% | AQZ41204.1, AQZ41205.1, CCF23512.1 |
| Buckfast: 4.76% | |||||
| Allele 28 | SSLSNKTIHNNNNYKYNYNNNNYNNNYNNNCKKLYYNIINIEQI | 44 | 1.45% | Mellifera: 25.00% | AEI99762.1, QEN96035.1 |
| Ligustica: 1.47% | |||||
| Allele 29 | SSLSNNYNYNNNNYNNNYNKKLYYNINYIEQI | 32 | 1.45% | Ligustica: 1.47% | ADJ57960.1, AEI99714.1, AEI99724.1, AEI99729.1, AEI99733.1, AEI99735.1, AEI99736.1, AEI99738.1, AEI99741.1, AEI99743.1, AEI99744.1, AEI99755.1, AEI99759.1 |
| Sicula: 12.50% | |||||
| Allele 30 | SSLSNNYNSNNYYNYNNNKKLYYKNYIINIEQI | 33 | 1.45% | Sicula: 25.00% | |
| Allele 31 | SSLSNKTIHNNNNYKYNYNNKYNYNNNNYNNNNYNKKLYYKNYIINIEQI | 50 | 2.17% | Sicula: 12.50% | AQZ41179.1, AQZ41181.1, AQZ41192.1, AQZ41193.1, QEN96025.1 |
| Ligustica: 2.94% | |||||
| Allele 32 | SSLSNSCNYSNNYNNNYNNTKKLYYNINYIEQI | 33 | 0.72% | Ligustica: 4.41% | AEI99783.1, AEI99788.1, AEI99792.1, AGZ61869.1 |
| Allele 33 | SSLSNKTIHNNNNYKNYNYKKLYYNIINIEQI | 32 | 0.72% | Ligustica: 1.47% | CCF23479.1 |
| Allele 34 | SSLSNNYNYSNYNNNNYKQLCYNINYIEQI | 30 | 0.72% | Ligustica: 1.47% | ABD14139.1, ABD14141.1, ABD14142.1, ABD14143.1, ABD14144.1, AGA84531.1, AGZ61875.1, QEN96094.1 |
| Allele 35 | SSLSNNYNYSNYNNYNNYNNNYNNYNNNYNNYKKLYYNINYIEQI | 45 | 0.72% | Buckfast: 2.38% | CCF23536.1 |
| Allele 36 | SSLSNNYNSNSYNNYNNNYYNNKKLQYYNINYIEQI | 36 | 0.72% | Ligustica: 1.47% | AGA84533.1, CCF23490.1, QEN96078.1 |
| Allele 37 | SSLSNKTIHNNNNYNNNNYNNYNNNNYNNYKKLYYNIINIEQI | 43 | 0.72% | Buckfast: 2.38% | QEN96087.1 |
| Allele 38 | SSLSSNYNSNNYNNYNNYKQLCYNINYIEQI | 31 | 0.72% | Mellifera: 25.00% | ART88596.1, CCF23477.1 |
| Allele 39 | SSLSNNYNYNNNKYNYNNNNYKQLCYNINYIEQI | 34 | 0.72% | Ligustica: 1.47% | AEI99717.1, AEI99718.1, AEI99720.1, AEI99721.1, AEI99726.1, AEI99732.1, AEI99734.1, AEI99740.1, AEI99742.1, AGA84529.1 |
| Allele 40 | SSLSNKTIHNNNNYNNNNYNNYNNNNYNNYKKLYYNINYIEQI | 43 | 0.72% | Ligustica: 1.47% | CCF23531.1 |
| Allele 41 | SSLSNNYKYSNYNNYNNNNYNNNNYNNNSKKLYYNIINIEQI | 42 | 0.72% | Ligustica: 1.47% | AGA84526.1, CCF23524.1 |
| Allele 42 | SSLSNKTIHNNNNYNNNNYKKLQYYNINYIEQI | 33 | 0.72% | Ligustica: 1.47% | ABV56220.1, AQZ41221.1, QEN96101.1 |
| Allele 43 | SSLSNNYNYNNNNYNNYNNNYNNNYNKKLYYNINYIEQI | 39 | 0.72% | Ligustica: 1.47% | QEN96052.1, QEN96059.1 |
| Allele 44 | SSLSNKTIHNNNYKYNYYNNNNYKKLQYYNIINIEQI | 37 | 0.72% | Ligustica: 1.47% | |
| Allele 45 | SSLSNNYNYNNNNYNNYNNYNNYNNNYNKKLYYNINYIEQI | 41 | 0.72% | Ligustica: 1.47% | ABV56219.1 |
| Allele 46 | SSLSNNYKYSNYNNYNNYNKKLYYKNYIINIEQI | 34 | 0.72% | Ligustica: 1.47% | ABD14104.1, ADJ57958.1, AGA84523.1, QEN96085.1, QEN96088.1 |
| Allele 47 | SSLSNNYNYNNNNYNNYNNYNNNYNNNYNKKLYYNINYIEQI | 42 | 0.72% | Ligustica: 1.47% | |
| Allele 48 | SSLSNNYKYSNYNNNNYNNNSKKLYYNINYIEQI | 34 | 0.72% | Ligustica: 1.47% | AQZ41199.1, CCF23485.1 |
| Allele 49 | SSLSNKTIHNNNNYNNNNYNKKLYYNINYIEQI | 33 | 0.72% | Ligustica: 1.47% | |
| Allele 50 | SSLSNKTIHNNNNYKYNYNNNNYNNNYNNYKKLYYNINYIEQI | 43 | 0.72% | Ligustica: 1.47% | CCF23532.1, QEN96032.1, QEN96033.1, QEN96042.1 |
| Allele 51 | SSLSNKTIHNNNNYKNYNNYKNYNNYKKLYYNINYIEQI | 39 | 0.72% | Carnica: 10.00% | CCF23513.1 |
| Allele 52 | SSLSNKTIHNNNNYKYNYNNNNYNNNNYNKKLYYKNYIINIEQI | 44 | 0.72% | Ligustica: 1.47% | CCF23527.1 |
| Allele 53 | SSLSNNTIHNNNYKYNYNNKYNYNNKKLYYNIINIEQI | 38 | 0.72% | Carnica: 10.00% | CCF23510.1, QEN96012.1 |
| Allele 54 | SSLSNKTIHNNNNYKYNYNNNNYNNNNYNNNYNNNCKKLYYNIINIEQI | 49 | 0.72% | Buckfast: 2.38% | AAQ57659.1, QEN96069.1 |
| Allele 55 | SSLSNNYNSNSYNNNYNNNYYNKKLQYYNINYIEQI | 36 | 0.72% | Buckfast: 2.38% | CCF23486.1 |
| Allele 56 | SSLSNNYKYSNYNNYNNYNNNNYNNYNNYNNKKLYYNIINIEQI | 44 | 0.72% | Buckfast: 2.38% | |
| Allele 57 | SSLSNKTIHNNNNYKKLYYNINYIEQI | 27 | 0.72% | Buckfast: 2.38% | CCF23466.1 |
| Allele 58 | SSLSNNYNYSNYNNYNNYKKLYYNINYIEQI | 31 | 0.72% | Buckfast: 2.38% | CCF23475.1 |
| Allele 59 | SSLSNNTIHNNNYKYNYNNNNYNNNNYNKKLYYNIINIEQI | 41 | 0.72% | Buckfast: 2.38% | ABD14102.1, ABD14103.1, AEI99760.1, AEI99784.1 |
| Allele 60 | SSLSNNYNYSNYNNYNNNNNYNNYKKLYYNINYIEQI | 37 | 0.72% | Ligustica: 1.47% | ABD14109.1, ABD14110.1, ABD14111.1, ABD14112.1, ABD14113.1, ABD14114.1, ABD14115.1, ABD14116.1, AQZ41223.1, QEN96102.1 |
| Allele 61 | SSLSNNYKYSNYNNYNNNYNNYNNNYKKLYYNINYIEQI | 39 | 0.72% | Ligustica: 1.47% | AEI99754.1, AEI99786.1, AEI99787.1, AEI99789.1 |
| Allele 62 | SSLSSSCNYSNNYNNYYNNNKKLYYNIINIEQI | 33 | 0.72% | Ligustica: 1.47% | AGA84525.1, AIS73042.1, AIS73050.1, CCF23484.1, QEN96007.1, QEN96009.1, QEN96018.1, QEN96019.1, QEN96026.1, QEN96061.1, QEN96074.1, QEN96084.1, QEN96097.1, QEN96105.1 |
| Allele 63 | SSLSNKTIHNNNKYNYNNNYNNNCKKLYYNINYIEQI | 37 | 0.72% | Cecropia: 50.00% | |
| Allele 64 | SSLSNNRNSNNYNNYNYKKLYYNINYIEQI | 30 | 0.72% | Buckfast: 2.38% | ABV56222.1, CCF23473.1 |
| Allele 65 | SSLSNNYNYSNYNNYNNNYNNNYNNNDYKKLYYKNYIINIEQI | 43 | 0.72% | Buckfast: 2.38% | ABV56216.1, QEN96106.1 |
| Allele 66 | SSLSNNYNYSNYNNYNNNNYNNYKKLYYNINYIEQI | 36 | 0.72% | Hyb. Carn: 25.00% | CCF23496.1 |
| Allele 67 | SSLSNNYNYSNNYNNYYNNNNNYNNYKKLYYNIINIEQI | 39 | 0.72% | Hyb. Carn: 25.00% | AEI99727.1, AEI99728.1 |
| Allele 68 | SSLSKNTIHNNNYNNSKKLYYNIINIEQI | 29 | 0.72% | Buckfast: 2.38% | AEI99716.1 |
| Allele 69 | SSLSNKTIHNNNNYNNNYNNNCKKLYYNIINIEQI | 35 | 0.72% | Mellifera: 25.00% | |
| Allele 70 | SSLSNKTIHNNNNYNNNNYNNNNYNNNNYKKLQYYNINYIEQI | 43 | 0.72% | Mellifera: 25.00% | |
| Allele 71 | SSLSNNYKYSNYNNYNNNNYKKLQYYNINYIEQI | 34 | 0.72% | Ligustica: 1.47% | |
| Allele 72 | SSLSNKTIHNNNNYKYNYNNNNYKPYYNINYIEQI | 35 | 0.72% | Ligustica: 1.47% | AEI99745.1 |
| Allele 73 | SSLSNKTIHNNNNYKYNYNNNYKKLYYKNYIINIEQI | 37 | 0.72% | Sicula: 12.50% | |
| Allele 74 | SSLSNKTIHNNNNYKYNYNNNYNNNSKKLQYYYNINYIEQI | 41 | 0.72% | Sicula: 12.50% | |
| Allele 75 | SSLSNKTIHNNNYKYNYNNKHNYNKLYYNINYIEQI | 36 | 0.72% | Sicula: 12.50% | |
| Allele 76 | SSLSNNYKYSNYNNYNNYNNNSKKLYKNYIINIEQI | 36 | 0.72% | Sicula: 12.50% | AQZ41187.1, AQZ41197.1, AQZ41198.1, ART88598.1, CCF23491.1, QEN96020.1, QEN96040.1 |
| Allele 77 | SSLSNKTIHNNNNYNNNNYNNYNNNNYNYKKLYYNINYIEQI | 42 | 0.72% | Ligustica: 1.47% | CCF23528.1 |
| Allele 78 | SSLSNKTIHNNNNYKYNYNNNYNNNSKKLYYNINYIEQI | 39 | 0.72% | Ligustica: 1.47% | AGA84537.1, CCF23514.1 |
| Allele 79 | SSLSNNYNYSNYNNYNNNYNNYNKKLYYNINYIEQI | 36 | 0.72% | Buckfast: 2.38% | ABD14117.1 |
| Allele 80 | SSLSNKTIHNNNNYKNYNNYKNYNNYKNYNNYKKLYYNINYIEQI | 45 | 0.72% | Buckfast: 2.38% | |
| Allele 81 | SSLSNNYNYNNYNNTNNINKQLYYNINYIEQI | 32 | 0.72% | Buckfast: 2.38% | ABV56218.1, AQZ41218.1, QEN96077.1, QEN96095.1 |
| Allele 82 | SSLSNNYSYNNYNNNNYNKKLYYNINYIEQI | 31 | 0.72% | Buckfast: 2.38% | CCF23476.1, QEN96068.1 |
| Allele 83 | SSLSNNYNYNNNNYNNYNNNYNKKLYYNINYIEQI | 35 | 0.72% | Buckfast: 2.38% | AQZ41215.1, AQZ41216.1, AQZ41217.1, CCF23487.1 |
| Allele 84 | SSLSNNYNYSNYNNYNNNNNYNNNNYNYKKLYYNINYIEQI | 41 | 0.72% | Buckfast: 2.38% | |
| Allele 85 | SSLSNKTIHNNNNNYNNYNKKLYYNIINIEQI | 32 | 0.72% | Ligustica: 1.47% | |
| Allele 86 | SSLSNNTIHNNNNYKYNYNNNYNNYNNYNNKKLYYNIINIEQI | 43 | 0.72% | Buckfast: 2.38% | CCF23529.1 |
| Allele 87 | SSLSTNTIHNNNNYKYNYNNNYNNYNNKKLYYNINYIEQI | 40 | 0.72% | Ligustica: 1.47% | |
| Allele 88 | SSLSNNYISNISNYNNNNNYNKKLYYNINYIEQI | 34 | 0.72% | Ligustica: 1.47% | AAQ67418.1, ABD14119.1, DAA06292.1, QEN96014.1, QEN96029.1, QEN96031.1, QEN96036.1, QEN96038.1, QEN96045.1, QEN96055.1, QEN96057.1, QEN96079.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolillo, G.; De Iorio, M.G.; Filipe, J.F.S.; Riva, F.; Stella, A.; Gandini, G.; Pagnacco, G.; Lazzari, B.; Minozzi, G. Analysis of Complementary Sex-Determiner (csd) Allele Diversity in Different Honeybee Subspecies from Italy Based on NGS Data. Genes 2022, 13, 991. https://doi.org/10.3390/genes13060991
Paolillo G, De Iorio MG, Filipe JFS, Riva F, Stella A, Gandini G, Pagnacco G, Lazzari B, Minozzi G. Analysis of Complementary Sex-Determiner (csd) Allele Diversity in Different Honeybee Subspecies from Italy Based on NGS Data. Genes. 2022; 13(6):991. https://doi.org/10.3390/genes13060991
Chicago/Turabian StylePaolillo, Gianluigi, Maria Grazia De Iorio, Joel F. Soares Filipe, Federica Riva, Alessandra Stella, Gustavo Gandini, Giulio Pagnacco, Barbara Lazzari, and Giulietta Minozzi. 2022. "Analysis of Complementary Sex-Determiner (csd) Allele Diversity in Different Honeybee Subspecies from Italy Based on NGS Data" Genes 13, no. 6: 991. https://doi.org/10.3390/genes13060991
APA StylePaolillo, G., De Iorio, M. G., Filipe, J. F. S., Riva, F., Stella, A., Gandini, G., Pagnacco, G., Lazzari, B., & Minozzi, G. (2022). Analysis of Complementary Sex-Determiner (csd) Allele Diversity in Different Honeybee Subspecies from Italy Based on NGS Data. Genes, 13(6), 991. https://doi.org/10.3390/genes13060991