
Integrated Analysis of a Ferroptosis-Related LncRNA Signature for Evaluating the Prognosis of Patients with Colorectal Cancer

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preprocessing
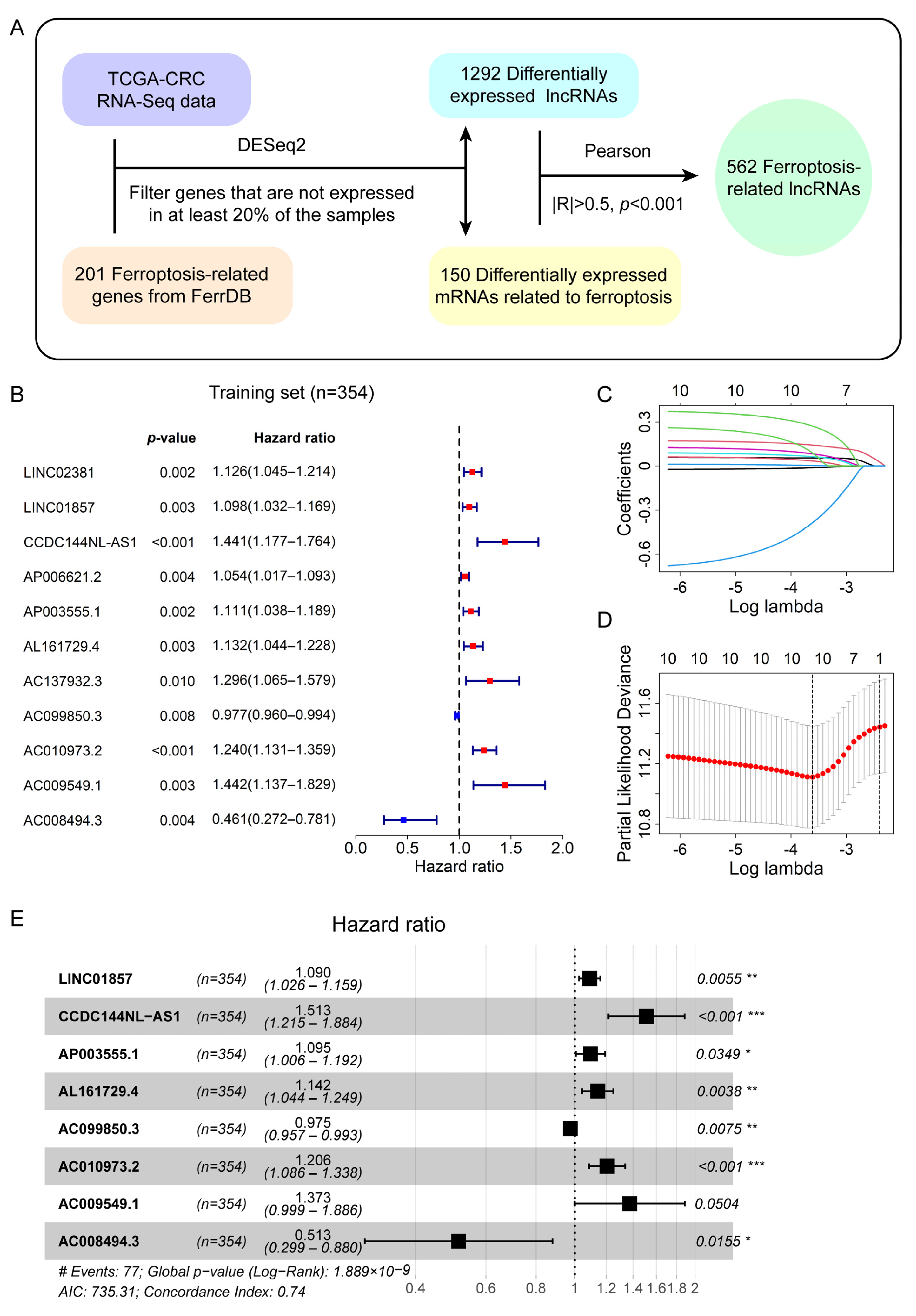
2.2. Identification of Differentially Expressed LncRNAs Related to Ferroptosis
2.3. Establishment of the Prognostic Signature Composed of Ferroptosis-Related LncRNAs in the Training Set
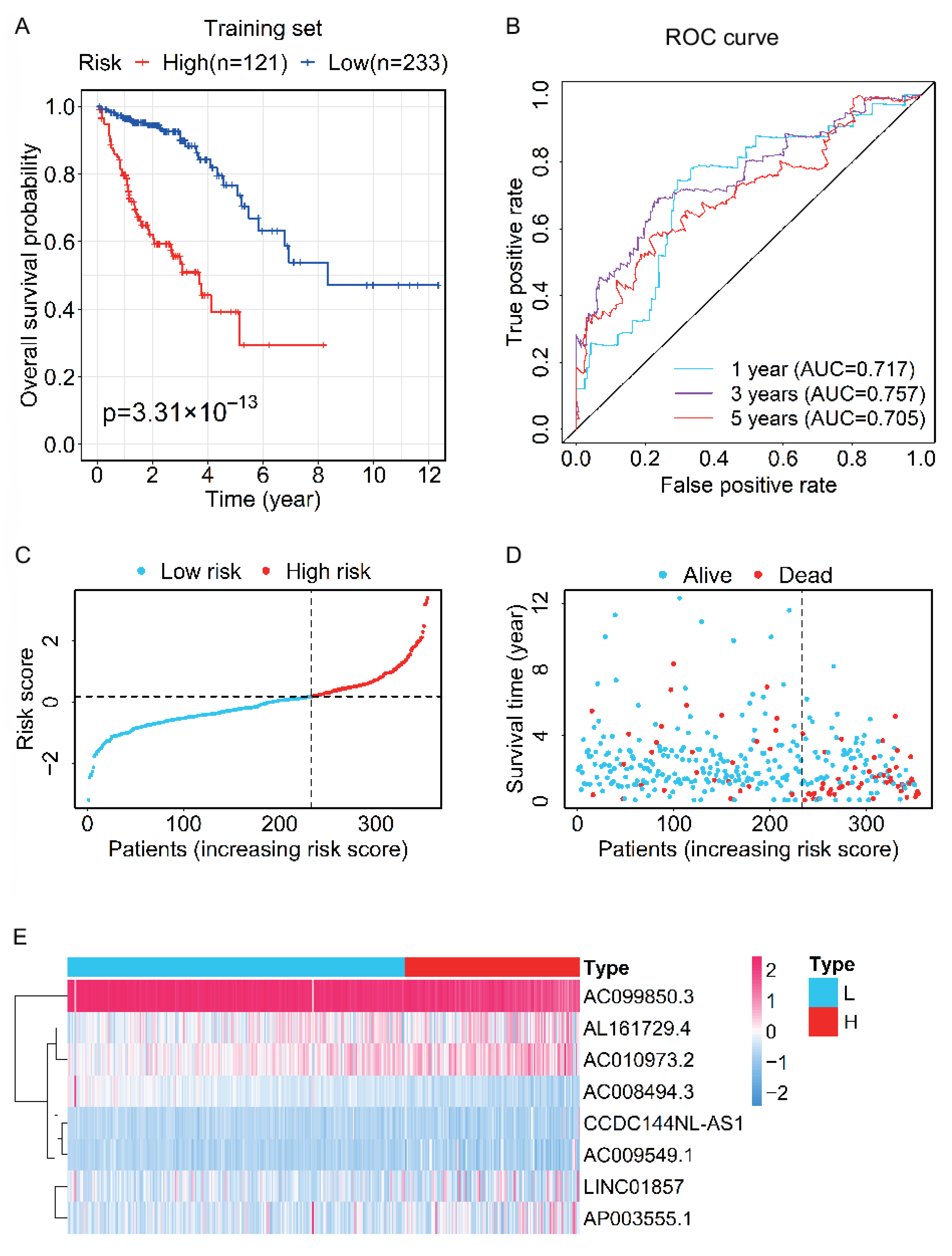
2.4. Evaluation and Validation of the Prognostic Capability for the Prognostic Signature in the Test and Entire Sets
2.5. The Relationship between Risk Score and Clinical Characteristics
2.6. Independent Prognostic Analysis of Risk Score and Construction of a Nomogram
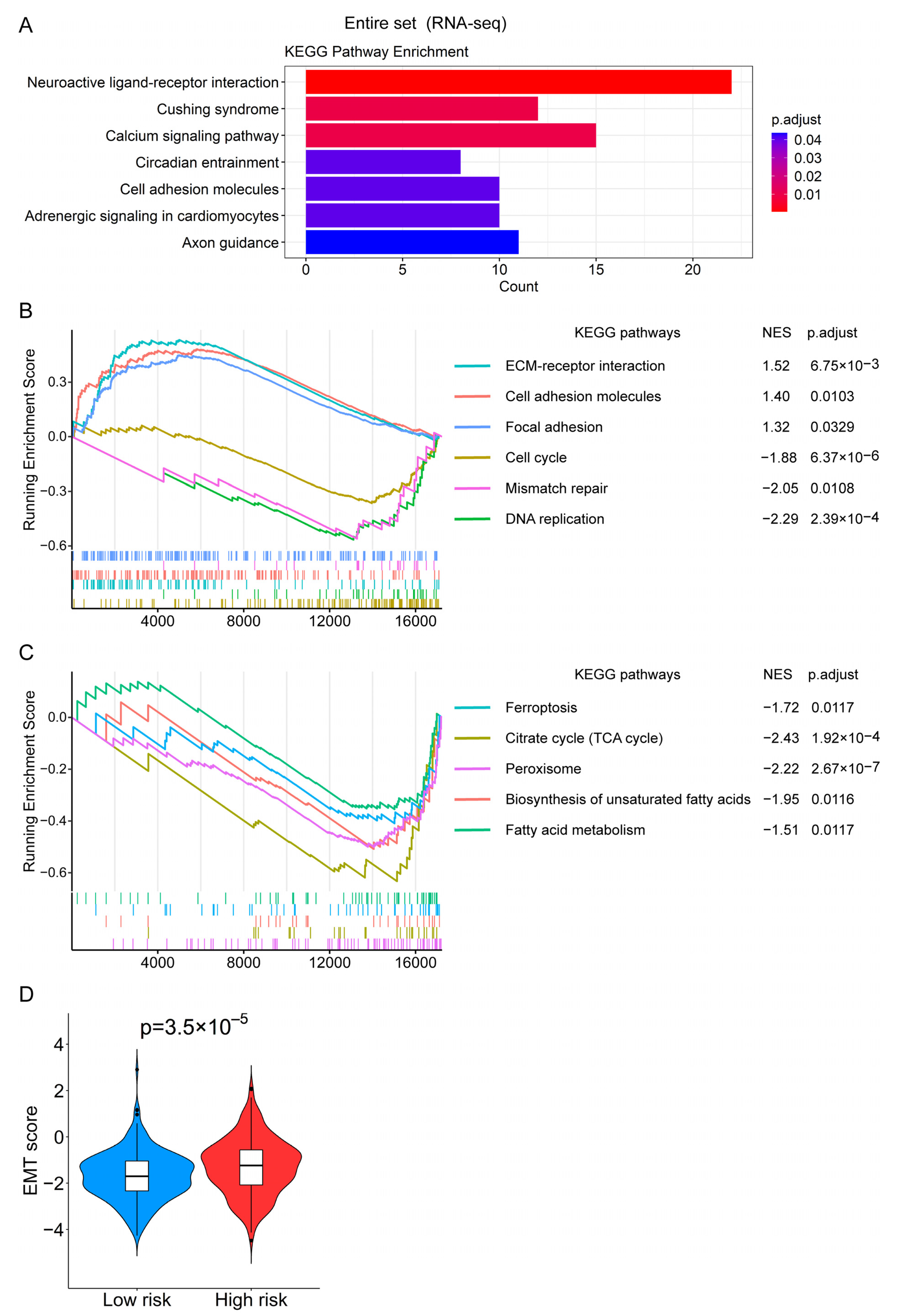
2.7. Functional Enrichment Analysis
2.8. Analysis of Somatic Mutation Data
2.9. Statistical Analysis
3. Results
3.1. Identification of Differentially Expressed Ferroptosis-Related LncRNAs in CRC Patients
3.2. A Prognostic Signature Consisting of Eight Ferroptosis-Related LncRNAs Was Derived from the Training Set
3.3. Evaluation and Validation of the Prognostic Performance for the Prognostic Signature
3.4. Analysis of the Relationship between the Prognostic Signature and the Clinical Characteristics of CRC Patients
3.5. The Prognostic Signature Can Be Applied as an Independent Prognostic Factor for CRC Patients
3.6. Construction and Verification of a Nomogram for the Prediction of the OS Rate
3.7. Functional Enrichment Analysis Revealed the Biological Processes Related to the Prognostic Signature
3.8. The Gene Mutation Profiling of CRC Patients between the High- and Low-Risk Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.P.; Kokudo, N.; Folprecht, G.; Mise, Y.; Unno, M.; Malik, H.Z.; Fenwick, S.W.; Poston, G.J. Colorectal Liver Metastases: A Critical Review of State of the Art. Liver Cancer 2016, 6, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Brodt, P.; Clavien, P.A.; Muschel, R.J.; D’Angelica, M.I.; Endo, I.; Parks, R.W.; Doyle, M.; de Santibanes, E.; Pawlik, T.M. Liver metastases. Nat. Rev. Dis. Primers 2021, 7, 27. [Google Scholar] [CrossRef]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Ahluwalia, P.; Kolhe, R.; Gahlay, G.K. The clinical relevance of gene expression based prognostic signatures in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188513. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, S.; Gong, X.; Tam, S.; Xiao, D.; Liu, S.; Tao, Y. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 2020, 19, 39. [Google Scholar] [CrossRef]
- Zou, Y.L.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Du, W.; Shi, X.; Chen, K.; Zhang, W.; Gao, M. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target. Ther. 2020, 5, 187. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yang, W.H.; Lin, Y.T.; Tang, X.; Chen, P.H.; Ding, C.C.; Qu, D.C.; Alvarez, J.V.; Chi, J.T. DDR2 upregulation confers ferroptosis susceptibility of recurrent breast tumors through the Hippo pathway. Oncogene 2021, 40, 2018–2034. [Google Scholar] [CrossRef] [PubMed]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespee, R.; Montero-Ruiz, L.; et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]
- Dai, E.Y.; Han, L.; Liu, J.; Xie, Y.C.; Zeh, H.J.; Kang, R.; Bai, L.L.; Tang, D.L. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 2020, 11, 6339. [Google Scholar] [CrossRef]
- Shen, L.D.; Qi, W.H.; Bai, J.J.; Zuo, C.Y.; Bai, D.L.; Gao, W.D.; Zong, X.L.; Hao, T.T.; Ma, Y.; Cao, G.C. Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat. Rec. 2021, 304, 313–322. [Google Scholar] [CrossRef]
- Song, J.; Liu, T.T.; Yin, Y.; Zhao, W.; Lin, Z.Q.; Yin, Y.X.; Lu, D.; You, F.P. The deubiquitinase OTUD1 enhances iron transport and potentiates host antitumor immunity. EMBO Rep. 2021, 22, e51162. [Google Scholar] [CrossRef]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; He, Y.; Lin, L.; Chen, P.; Chen, M.; Zhang, S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis 2021, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.P.; Kopetz, S.; Ajani, J.A.; Calin, G.A. Non-coding RNAs in GI cancers: From cancer hallmarks to clinical utility. Gut 2020, 69, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, J.; Luan, S.; He, C.; Li, Z. Long non-coding RNAs involved in cancer metabolic reprogramming. Cell Mol. Life Sci. 2019, 76, 495–504. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Liu, H.; Li, D.; Sun, L.; Qin, H.; Fan, A.; Meng, L.; Graves-Deal, R.; Glass, S.E.; Franklin, J.L.; Liu, Q.; et al. Interaction of lncRNA MIR100HG with hnRNPA2B1 facilitates m(6)A-dependent stabilization of TCF7L2 mRNA and colorectal cancer progression. Mol. Cancer 2022, 21, 74. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Xu, S.; Xie, J.; Zhou, Y.; Liu, H.; Wang, Y.; Li, Z. Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients. Genes 2022, 13, 345. [Google Scholar] [CrossRef]
- Qi, W.; Li, Z.; Xia, L.; Dai, J.; Zhang, Q.; Wu, C.; Xu, S. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci. Rep. 2019, 9, 16185. [Google Scholar] [CrossRef]
- Chen, B.; Wang, H.; Lv, C.; Mao, C.; Cui, Y. Long non-coding RNA H19 protects against intracerebral hemorrhage injuries via regulating microRNA-106b-5p/acyl-CoA synthetase long chain family member 4 axis. Bioengineered 2021, 12, 4004–4015. [Google Scholar] [CrossRef]
- Wei, X.B.; Jiang, W.Q.; Zeng, J.H.; Huang, L.Q.; Ding, H.G.; Jing, Y.W.; Han, Y.L.; Li, Y.C.; Chen, S.L. Exosome-Derived lncRNA NEAT1 Exacerbates Sepsis-Associated Encephalopathy by Promoting Ferroptosis Through Regulating miR-9-5p/TFRC and GOT1 Axis. Mol. Neurobiol. 2022, 59, 1954–1969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fang, D.; Li, S.; Bao, X.; Jiang, L.; Sun, X. Construction and Validation of a Novel Ferroptosis-Related lncRNA Signature to Predict Prognosis in Colorectal Cancer Patients. Front. Genet. 2021, 12, 709329. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2021, 12, 783362. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Huang, T.; Jin, M.; Zheng, Z.; Zhang, H.; Ye, M.; Liu, K. Establishment of a novel ferroptosis-related lncRNA pair prognostic model in colon adenocarcinoma. Aging 2021, 13, 23072–23095. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in S, R Package Version 2.37-7; Springer: New York, NY, USA, 2014.
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Harrell, F. rms: Regression Modeling Strategies; Springer International Publishing: New York, NY, USA, 2015. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Weng, Y.; Pan, Y.; Huang, Z.; Chen, X.; Hong, W.; Lin, T.; Wang, L.; Liu, W.; Qiu, S. A Novel Prognostic Signature Based on Metabolism-Related Genes to Predict Survival and Guide Personalized Treatment for Head and Neck Squamous Carcinoma. Front. Oncol. 2021, 11, 685026. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Brunson, J.C. ggalluvial: Layered Grammar for Alluvial Plots. J. Open Source Softw. 2020, 5, 2017. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Qian, L.; Jiang, S.; Gai, X.; Ye, S.; Chen, Y.; Wang, X.; Zhai, L.; Xu, J.; et al. A proteomic and phosphoproteomic landscape of KRAS mutant cancers identifies combination therapies. Mol. Cell 2021, 81, 4076–4090. [Google Scholar] [CrossRef]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef]
- Margonis, G.A.; Kim, Y.; Spolverato, G.; Ejaz, A.; Gupta, R.; Cosgrove, D.; Anders, R.; Karagkounis, G.; Choti, M.A.; Pawlik, T.M. Association Between Specific Mutations in KRAS Codon 12 and Colorectal Liver Metastasis. JAMA Surg. 2015, 150, 722–729. [Google Scholar] [CrossRef]
- Chun, Y.S.; Passot, G.; Yamashita, S.; Nusrat, M.; Katsonis, P.; Loree, J.M.; Conrad, C.; Tzeng, C.D.; Xiao, L.; Aloia, T.A.; et al. Deleterious Effect of RAS and Evolutionary High-risk TP53 Double Mutation in Colorectal Liver Metastases. Ann. Surg. 2019, 269, 917–923. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Yeh, Y.M.; Wu, P.Y.; Hsu, K.F.; Chang, J.Y.; Shen, M.R. Germline susceptibility variants impact clinical outcome and therapeutic strategies for stage III colorectal cancer. Sci. Rep. 2019, 9, 3931. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ge, X.; Tang, Y.; Qian, Y.; Lu, W.; Jiang, K.; Fang, Y.; Hwang, M.; Fu, D.; Xiao, Q.; et al. An Autophagy-Related Long Noncoding RNA Signature Contributes to Poor Prognosis in Colorectal Cancer. J. Oncol. 2020, 2020, 4728947. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Ge, Y.; Ma, X.; Li, Z.; Shi, L.; Lin, L.; Xiao, J.; Chen, W.; Ni, P.; Yang, L.; et al. Long non-coding RNA CCDC144NL-AS1 sponges miR-143-3p and regulates MAP3K7 by acting as a competing endogenous RNA in gastric cancer. Cell Death Dis. 2020, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Liu, N.; Wang, H.; Wang, Y.; Guo, Z. LncRNA LINC01857 promotes growth, migration, and invasion of glioma by modulating miR-1281/TRIM65 axis. J. Cell Physiol. 2019, 234, 22009–22016. [Google Scholar] [CrossRef]
- Li, Q.; Li, B.; Lu, C.L.; Wang, J.Y.; Gao, M.; Gao, W. LncRNA LINC01857 promotes cell growth and diminishes apoptosis via PI3K/mTOR pathway and EMT process by regulating miR-141-3p/MAP4K4 axis in diffuse large B-cell lymphoma. Cancer Gene Ther. 2020, 28, 1046–1057. [Google Scholar] [CrossRef]
- Huang, Q.Y.; Liu, G.F.; Qian, X.L.; Tang, L.B.; Huang, Q.Y.; Xiong, L.X. Long Non-Coding RNA: Dual Effects on Breast Cancer Metastasis and Clinical Applications. Cancers 2019, 11, 1802. [Google Scholar] [CrossRef]
- Ferris, R.L.; Galon, J. Additional Support for the Introduction of Immune Cell Quantification in Colorectal Cancer Classification. J. Natl. Cancer Inst. 2016, 108, djw033. [Google Scholar] [CrossRef][Green Version]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Griffith, L.; Wells, J.Z.; Taylor, D.P. The dormancy dilemma: Quiescence versus balanced proliferation. Cancer Res. 2013, 73, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, Y.D.; Yu, G.Y.; Cui, J.R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.G.; Deng, L.L.; Liu, P.; et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J., 3rd; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef]
- Tang, D.L.; Kroemer, G. Peroxisome: The new player in ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 273. [Google Scholar] [CrossRef]
- Cui, W.W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021, 28, 2536–2551. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Ensembl ID | Genomic Location (GRCh38) | Coefficient |
|---|---|---|---|
| LINC01857 | ENSG00000224137 | Chr2: 207,662,375–207,667,024 | 0.0865 |
| CCDC144NL-AS1 | ENSG00000233098 | Chr17: 20,868,433–21,002,276 | 0.4138 |
| AP003555.1 | ENSG00000254605 | Chr11: 70,014,858–70,021,059 | 0.0909 |
| AL161729.4 | ENSG00000271659 | Chr9: 95,514,045–95,514,520 | 0.1325 |
| AC099850.3 | ENSG00000265415 | Chr17: 59,202,677–59,203,829 | −0.0251 |
| AC010973.2 | ENSG00000244151 | Chr7: 151,074,742–151,076,530 | 0.187 |
| AC009549.1 | ENSG00000270607 | Chr11: 19,710,934–19,712,619 | 0.3169 |
| AC008494.3 | ENSG00000271797 | Chr5: 115,262,505–115,263,448 | −0.668 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Zhou, Y.; Luo, J.; Chen, S.; Xie, J.; Liu, H.; Wang, Y.; Li, Z. Integrated Analysis of a Ferroptosis-Related LncRNA Signature for Evaluating the Prognosis of Patients with Colorectal Cancer. Genes 2022, 13, 1094. https://doi.org/10.3390/genes13061094
Xu S, Zhou Y, Luo J, Chen S, Xie J, Liu H, Wang Y, Li Z. Integrated Analysis of a Ferroptosis-Related LncRNA Signature for Evaluating the Prognosis of Patients with Colorectal Cancer. Genes. 2022; 13(6):1094. https://doi.org/10.3390/genes13061094
Chicago/Turabian StyleXu, Shaohua, Yanjie Zhou, Junyun Luo, Su Chen, Jiahui Xie, Hui Liu, Yirong Wang, and Zhaoyong Li. 2022. "Integrated Analysis of a Ferroptosis-Related LncRNA Signature for Evaluating the Prognosis of Patients with Colorectal Cancer" Genes 13, no. 6: 1094. https://doi.org/10.3390/genes13061094
APA StyleXu, S., Zhou, Y., Luo, J., Chen, S., Xie, J., Liu, H., Wang, Y., & Li, Z. (2022). Integrated Analysis of a Ferroptosis-Related LncRNA Signature for Evaluating the Prognosis of Patients with Colorectal Cancer. Genes, 13(6), 1094. https://doi.org/10.3390/genes13061094